VM2: Computation of Binding Free Energies for Ligand Series Ranking
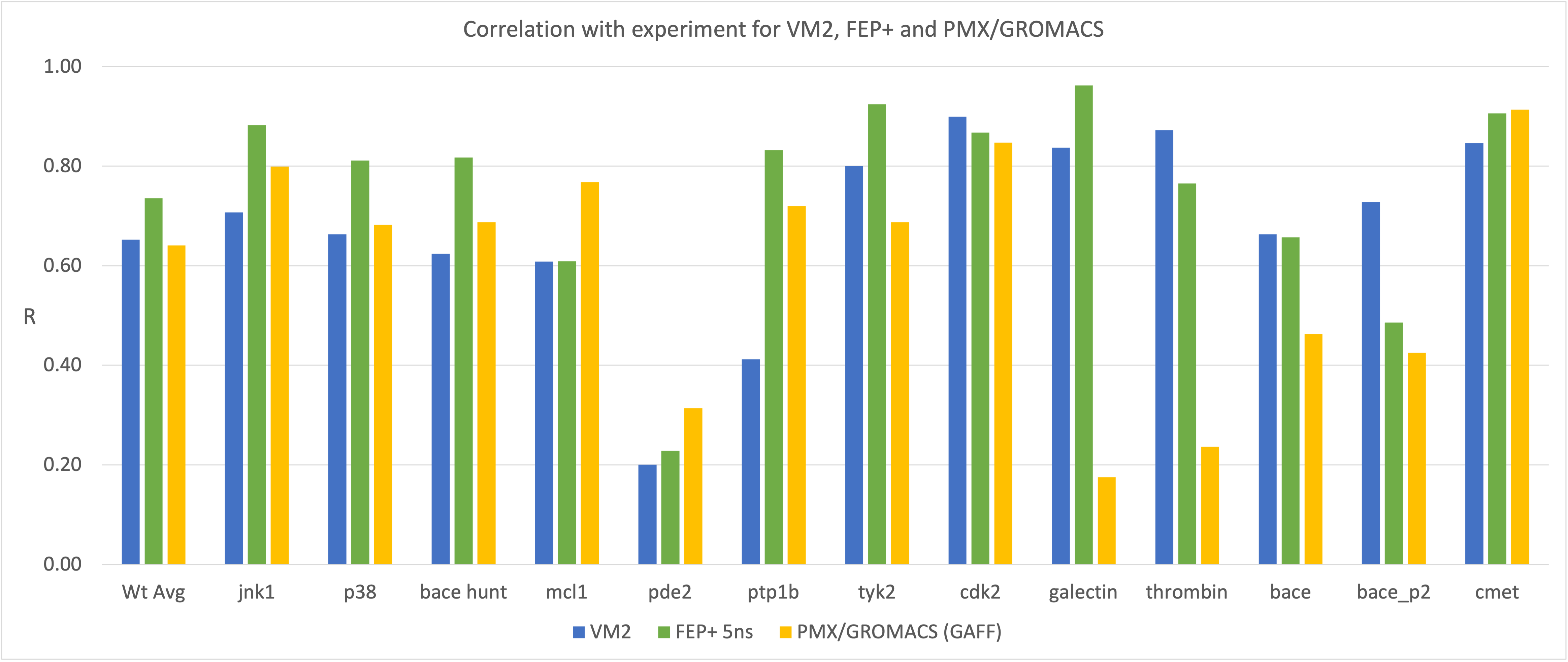
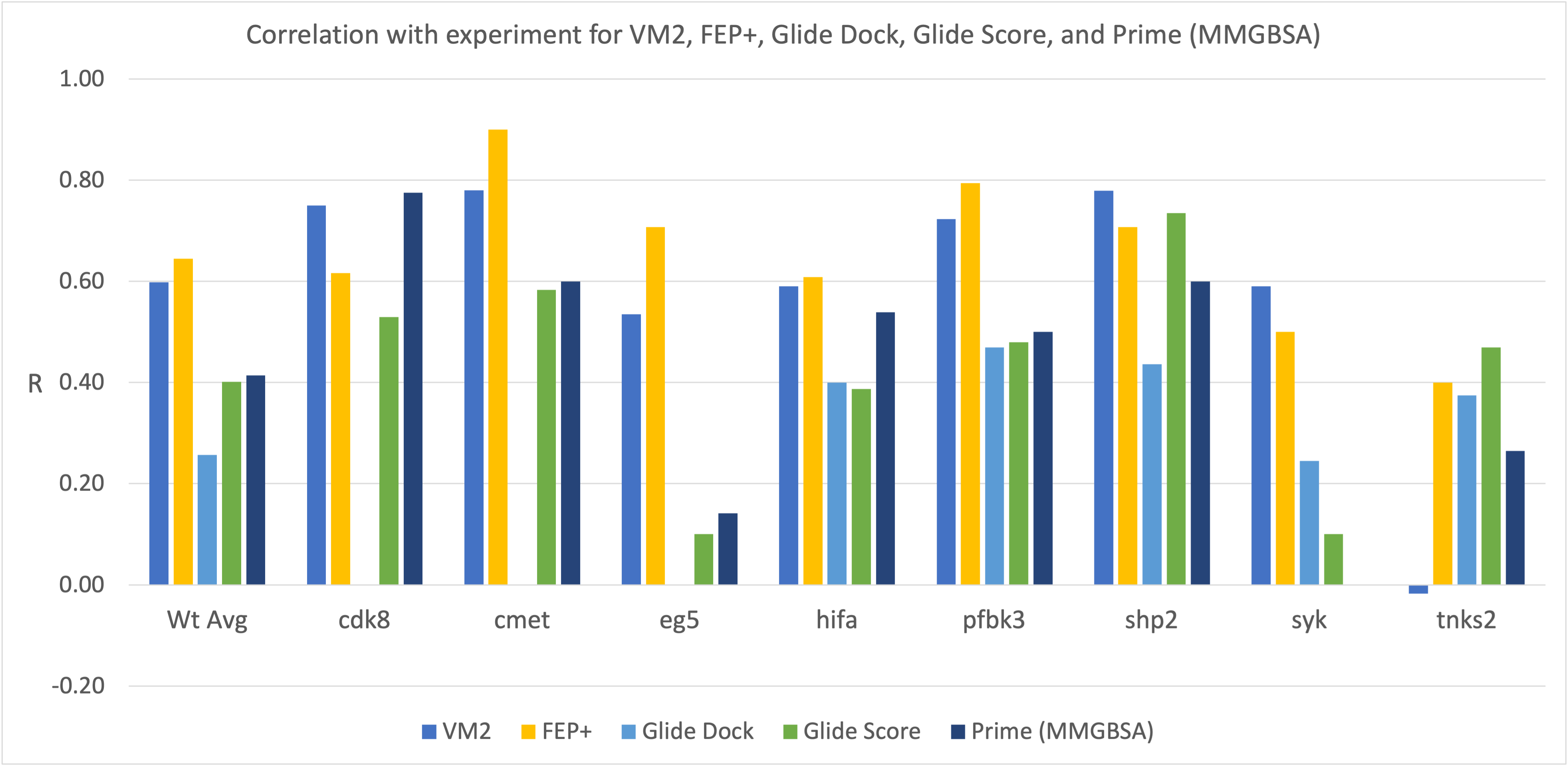
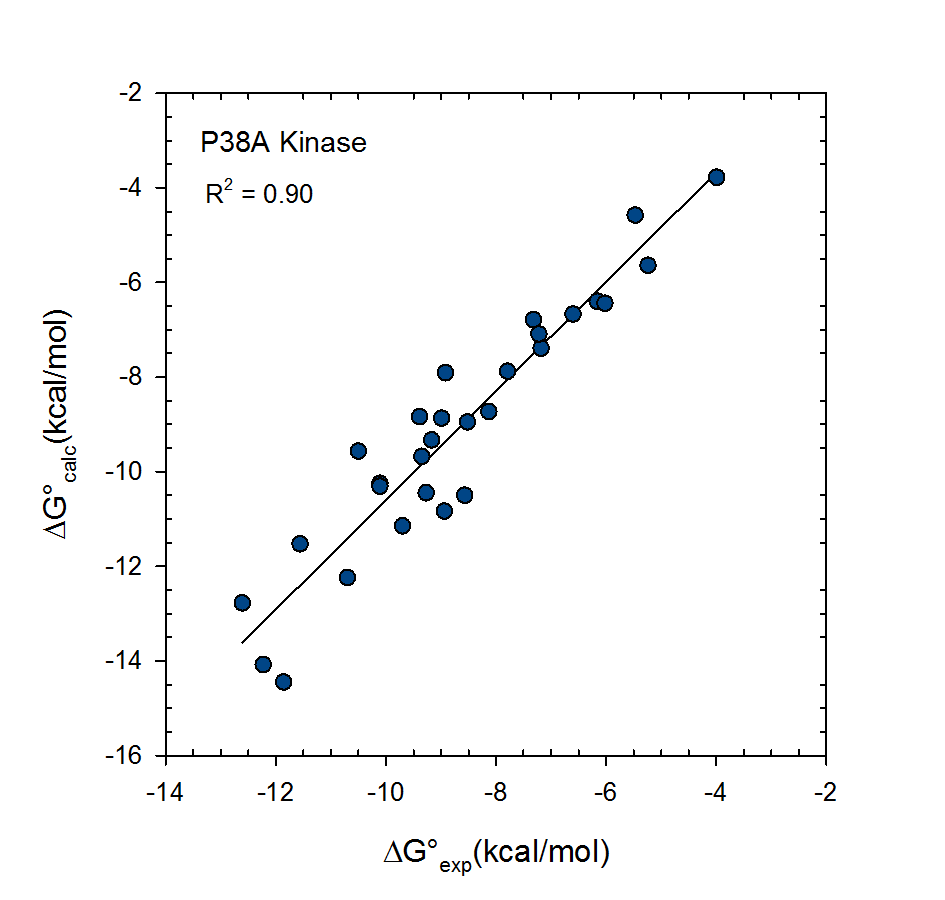
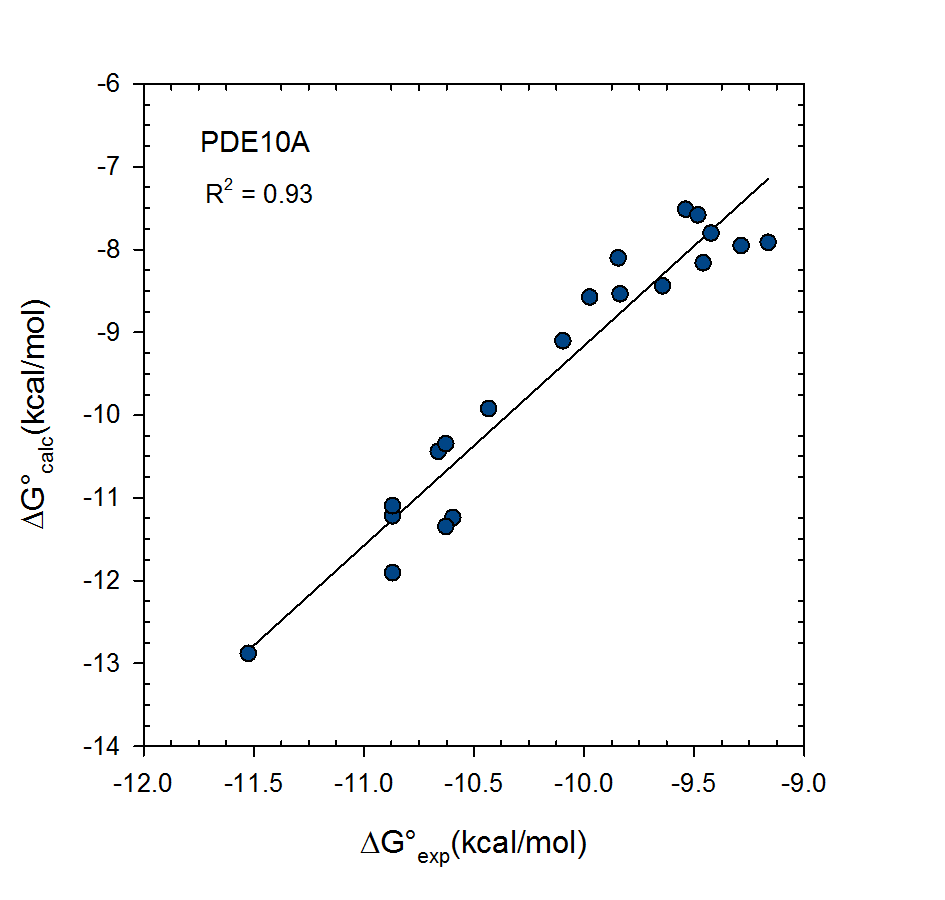
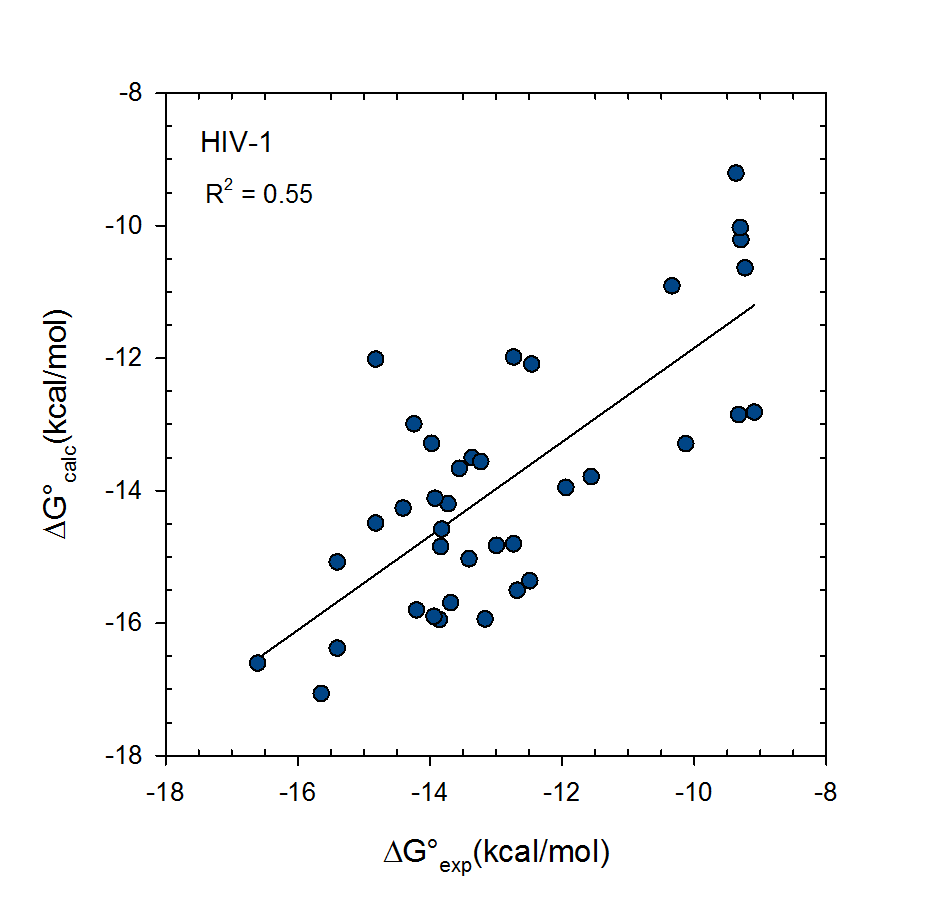
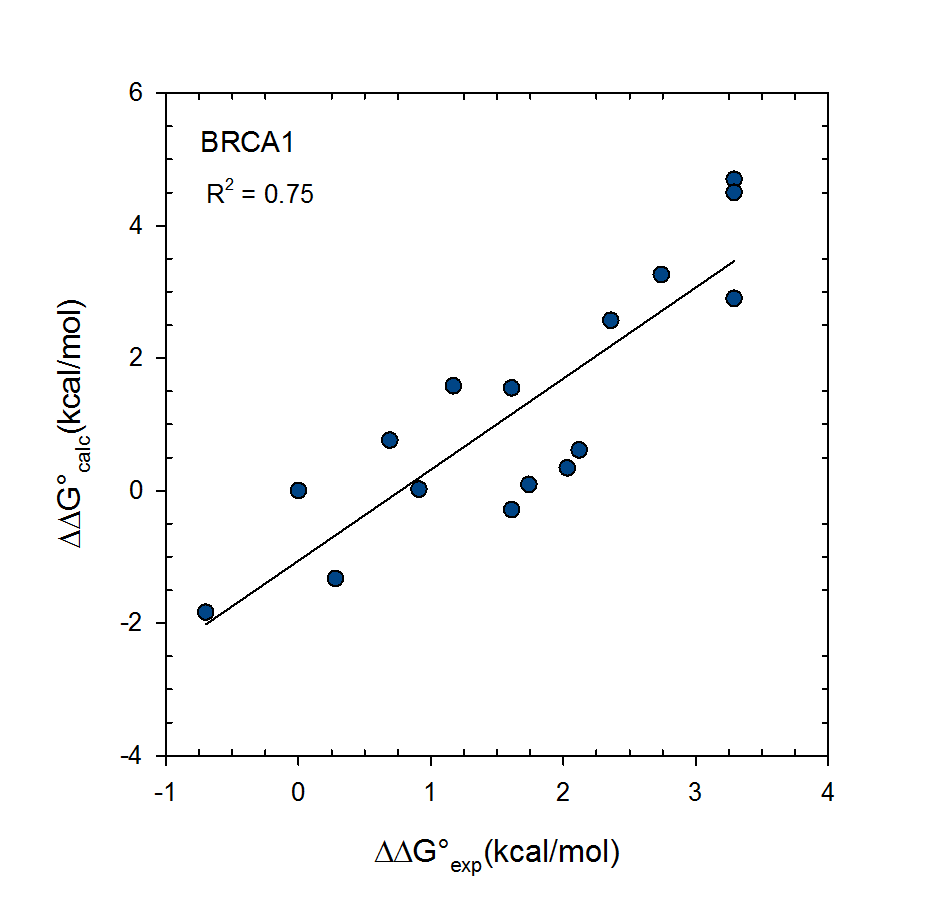
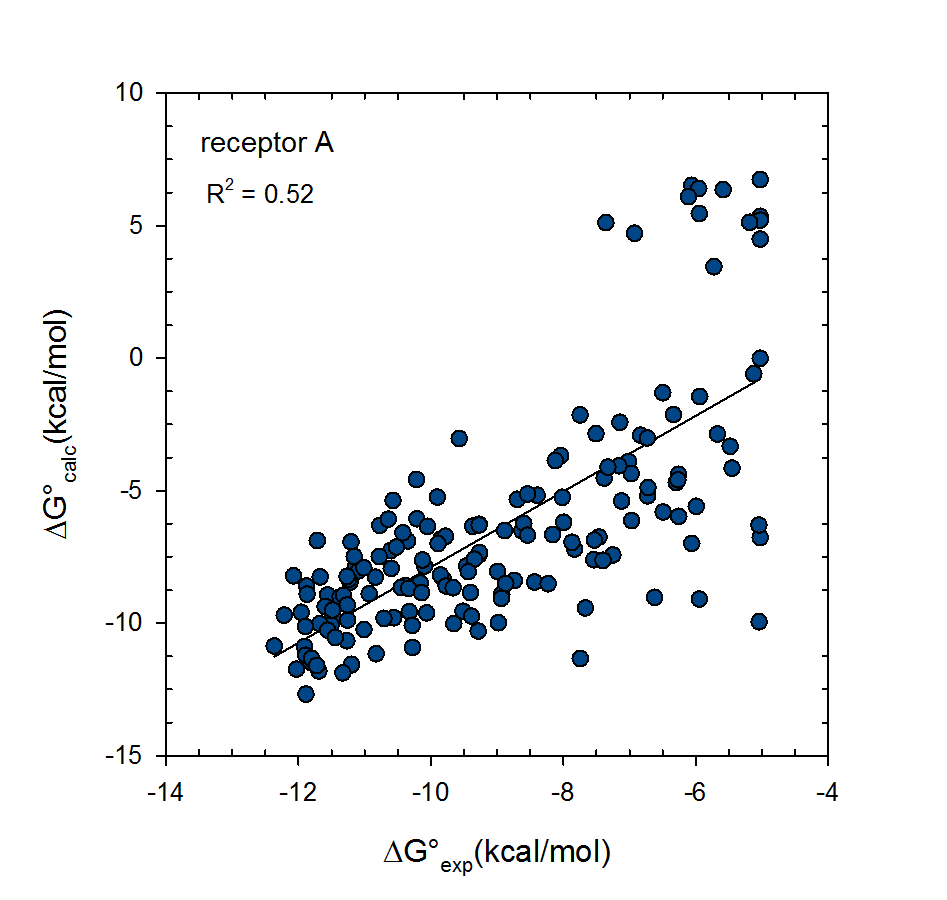
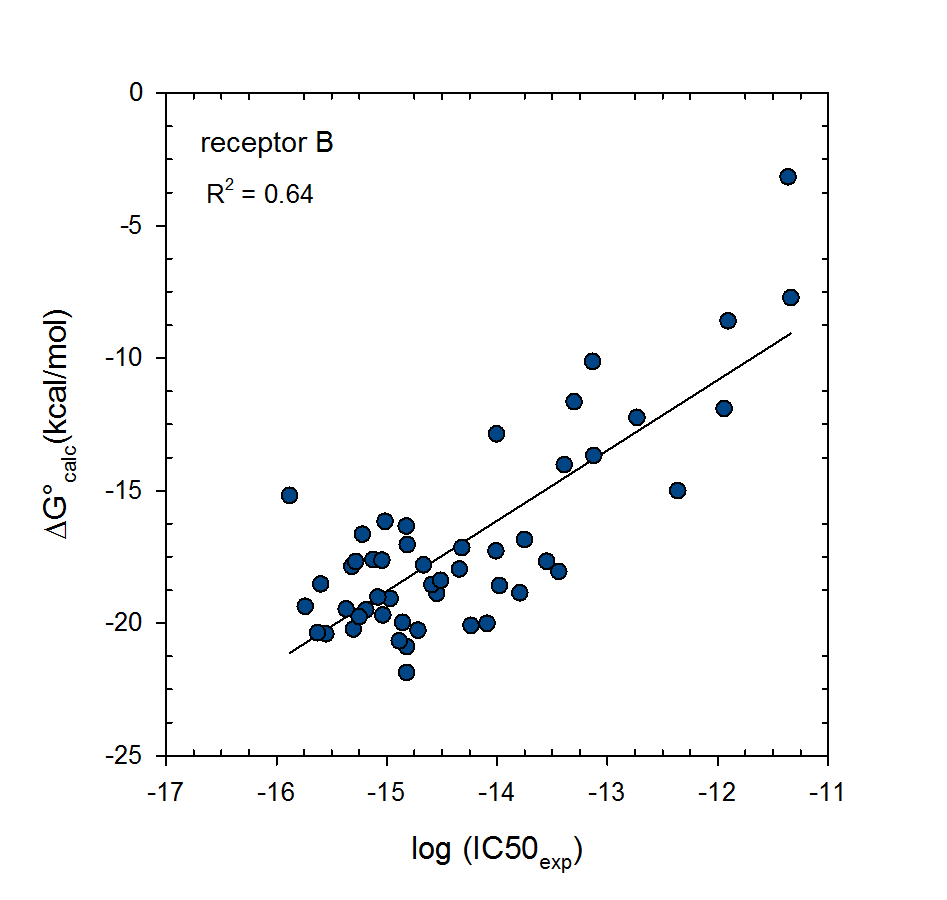
© 2007-2023 VeraChem, LLC
![]()