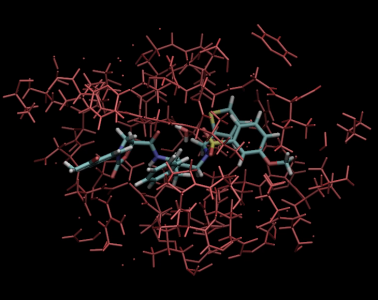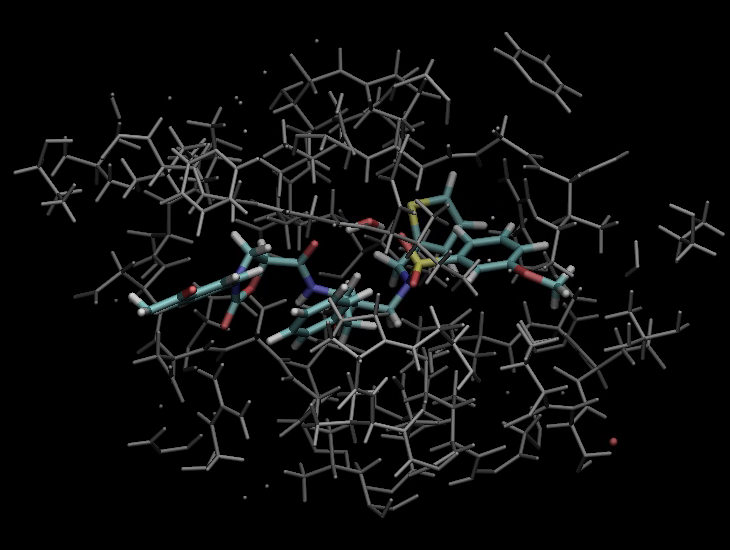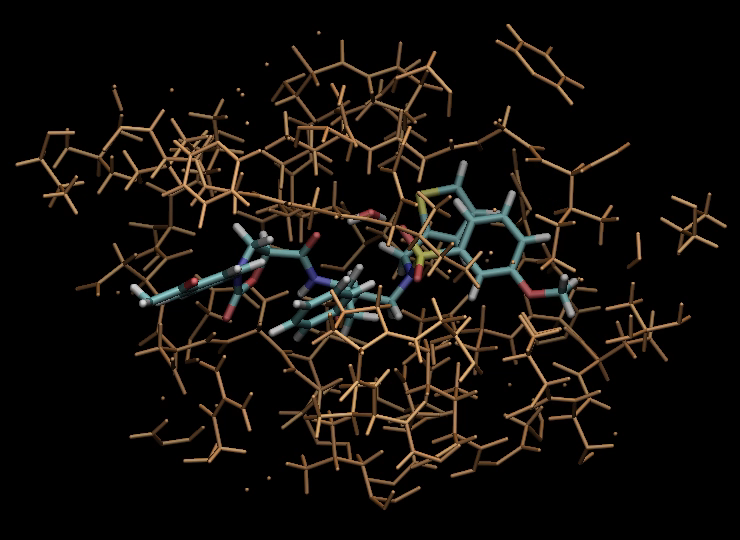
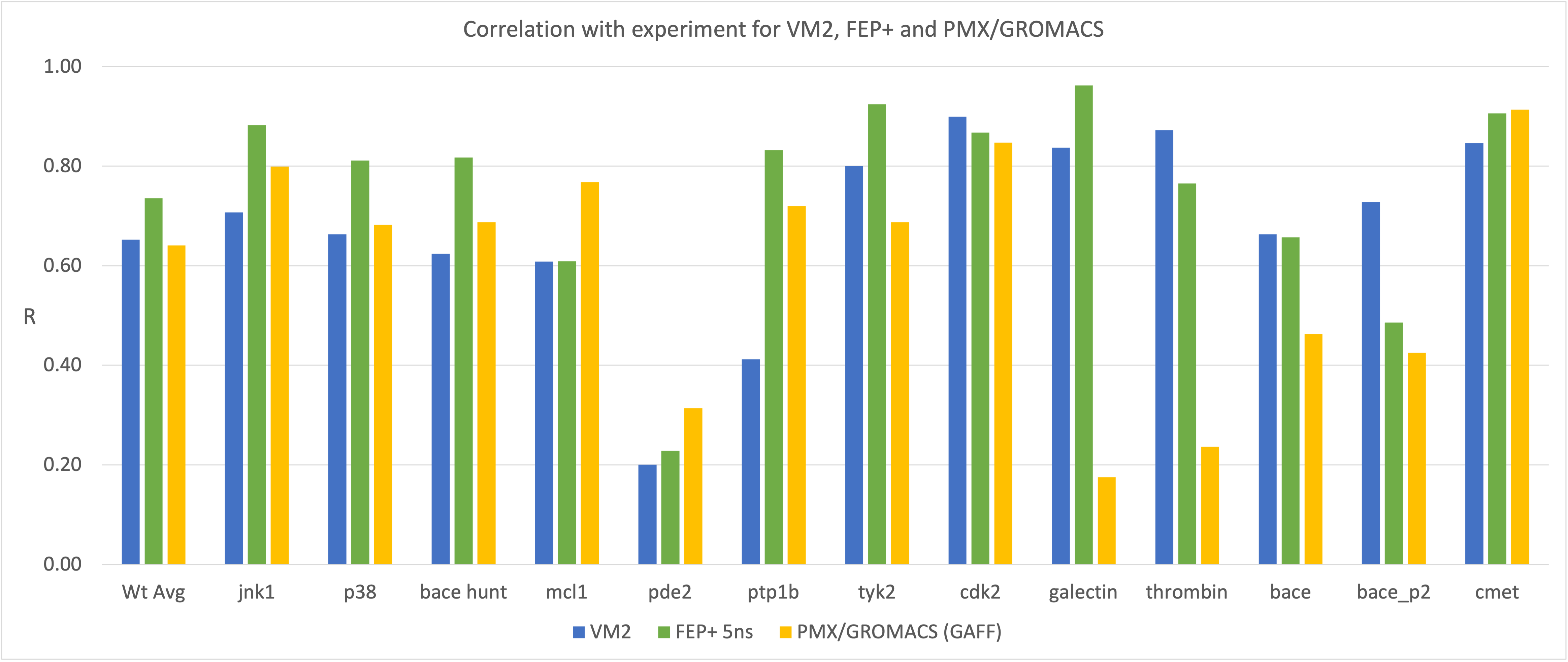
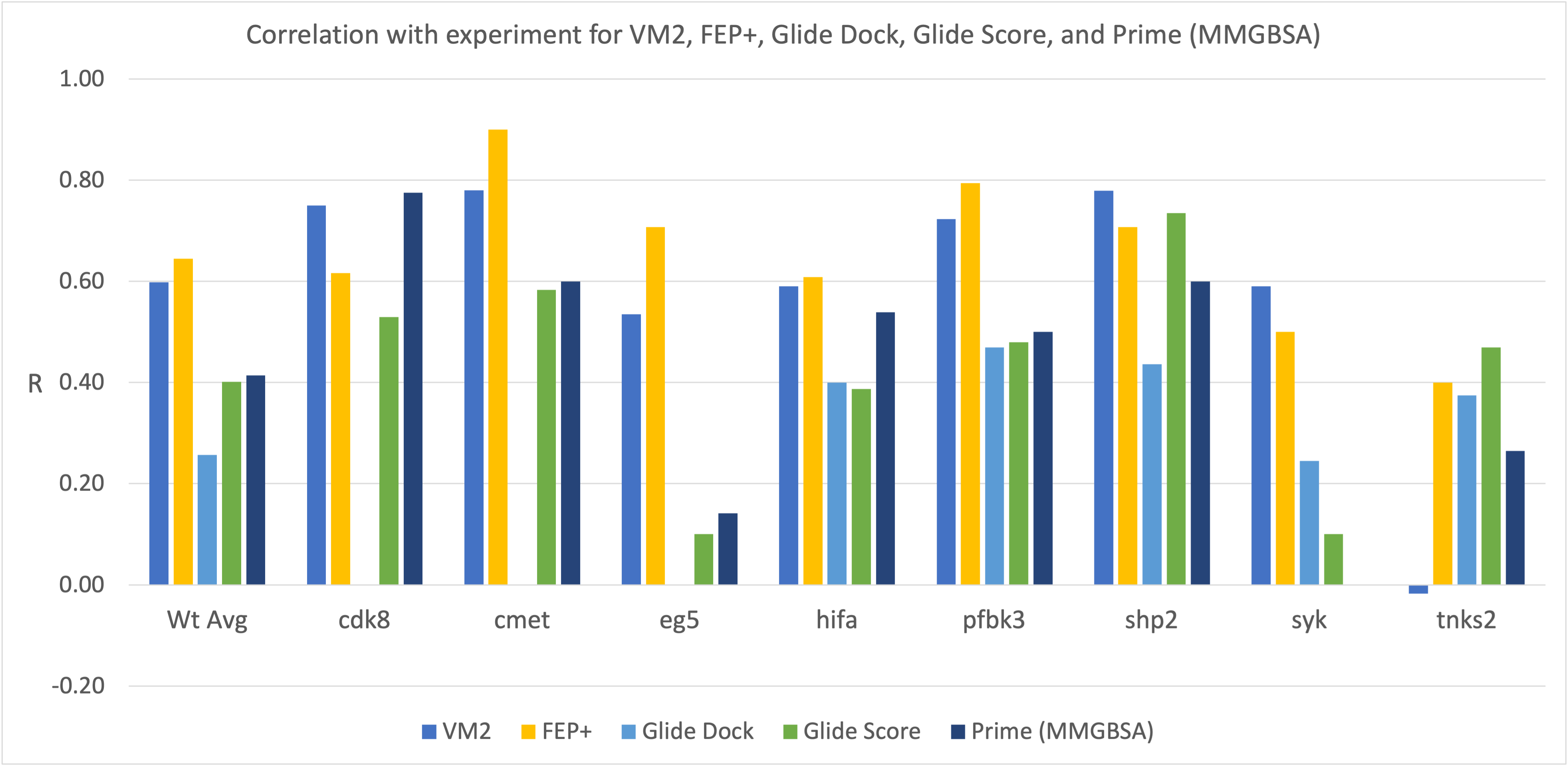
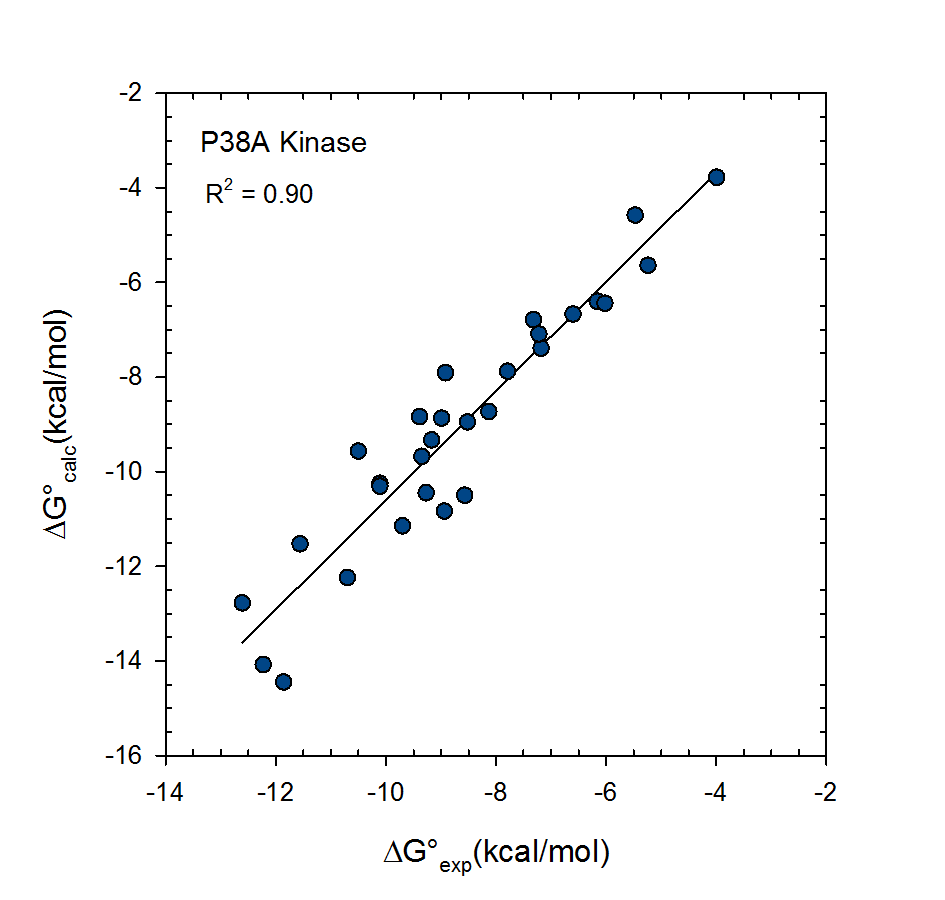
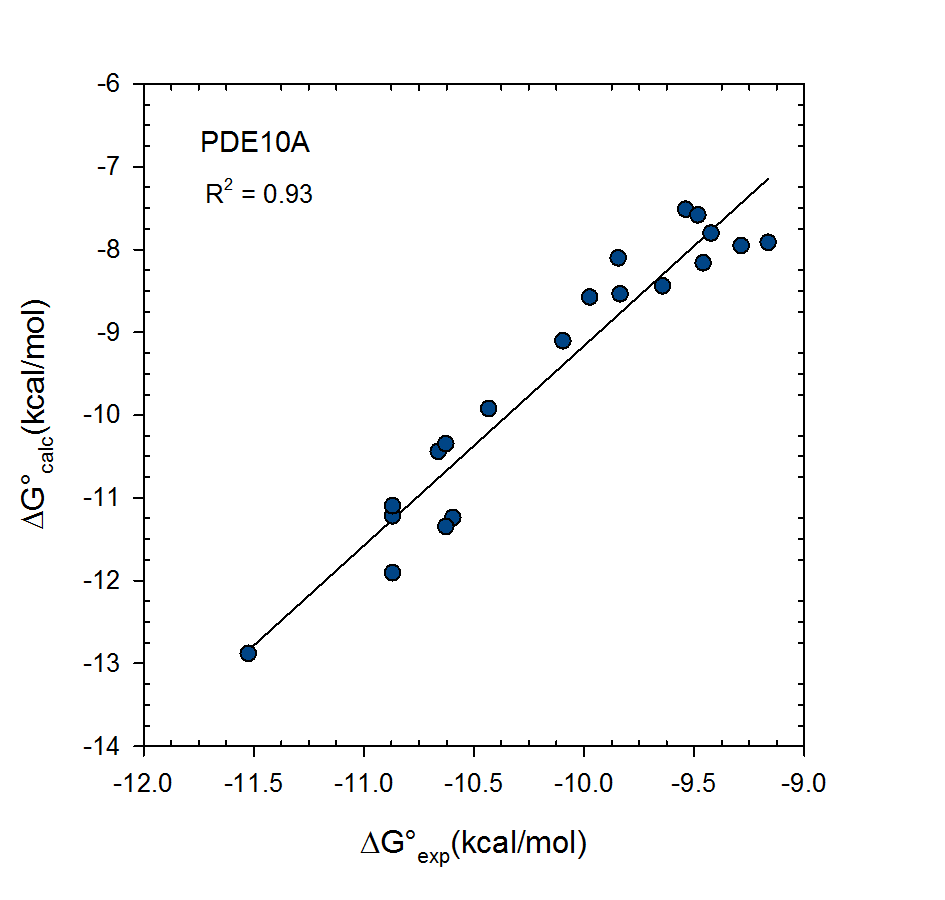
VM2: Free Energy Calculations by Mining Minima
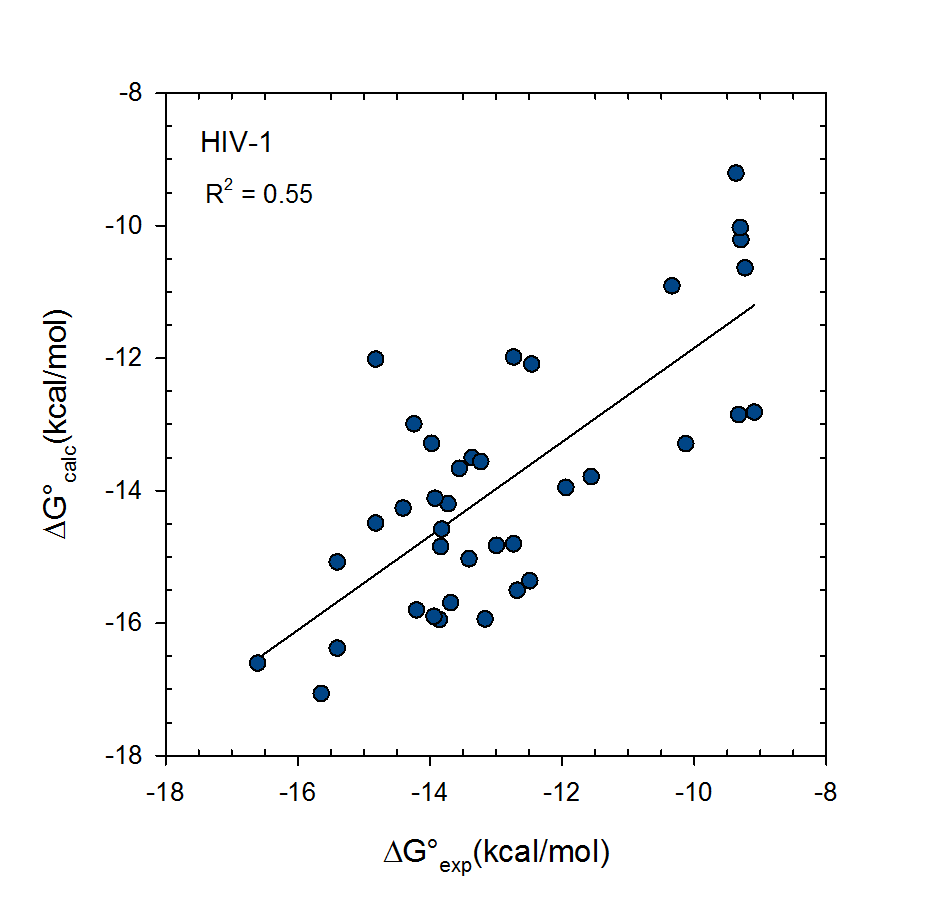
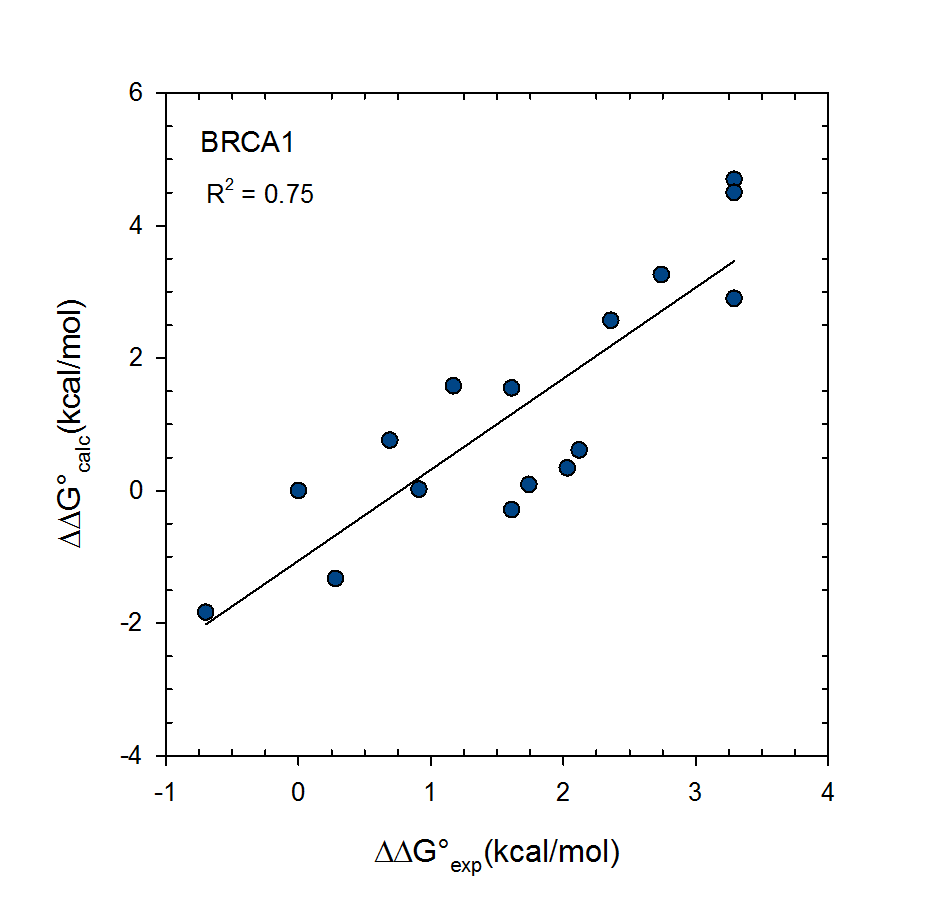
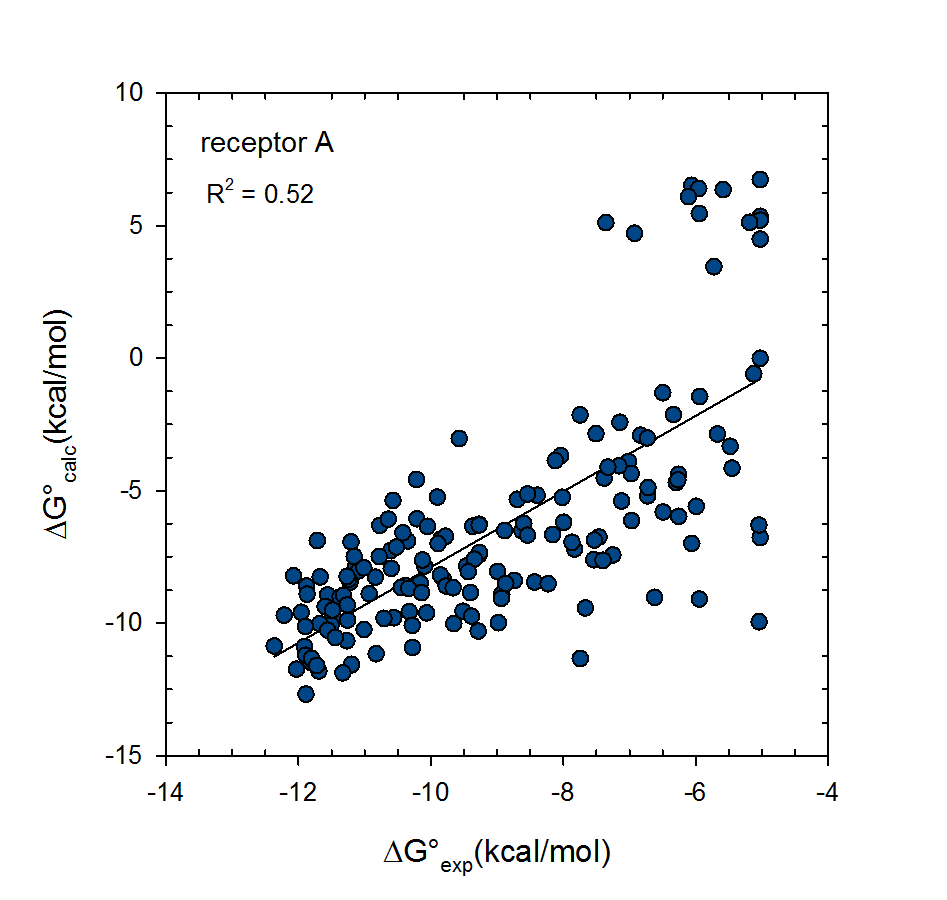
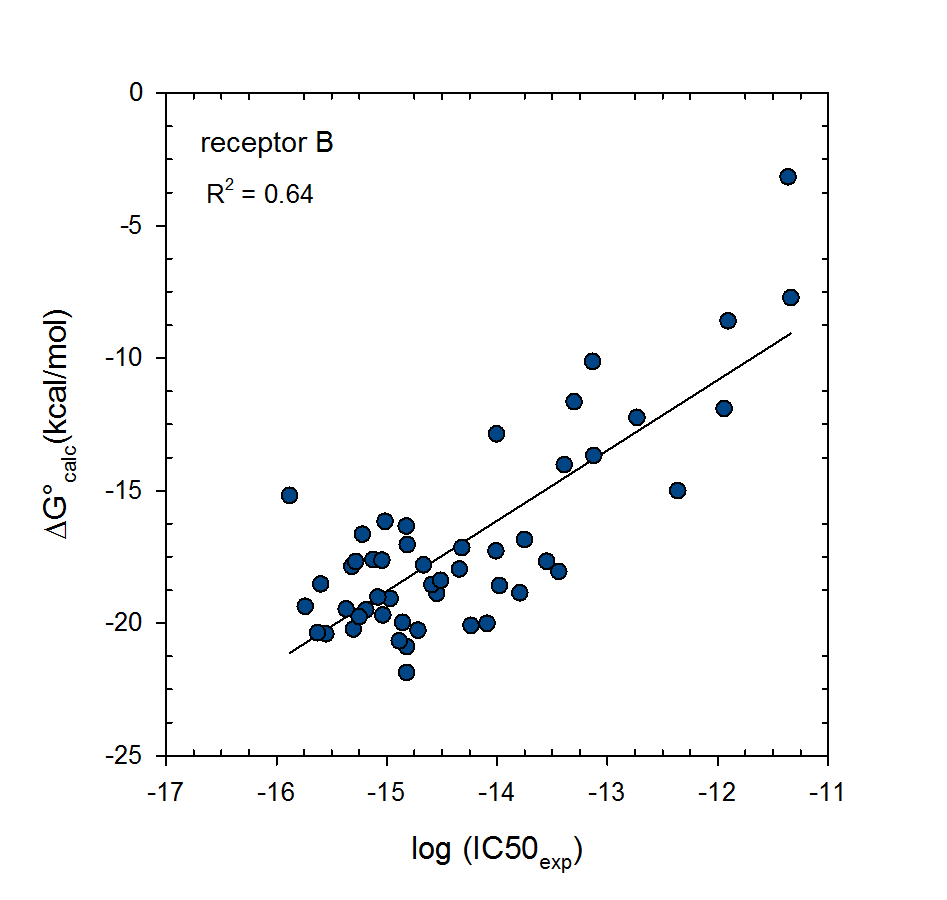
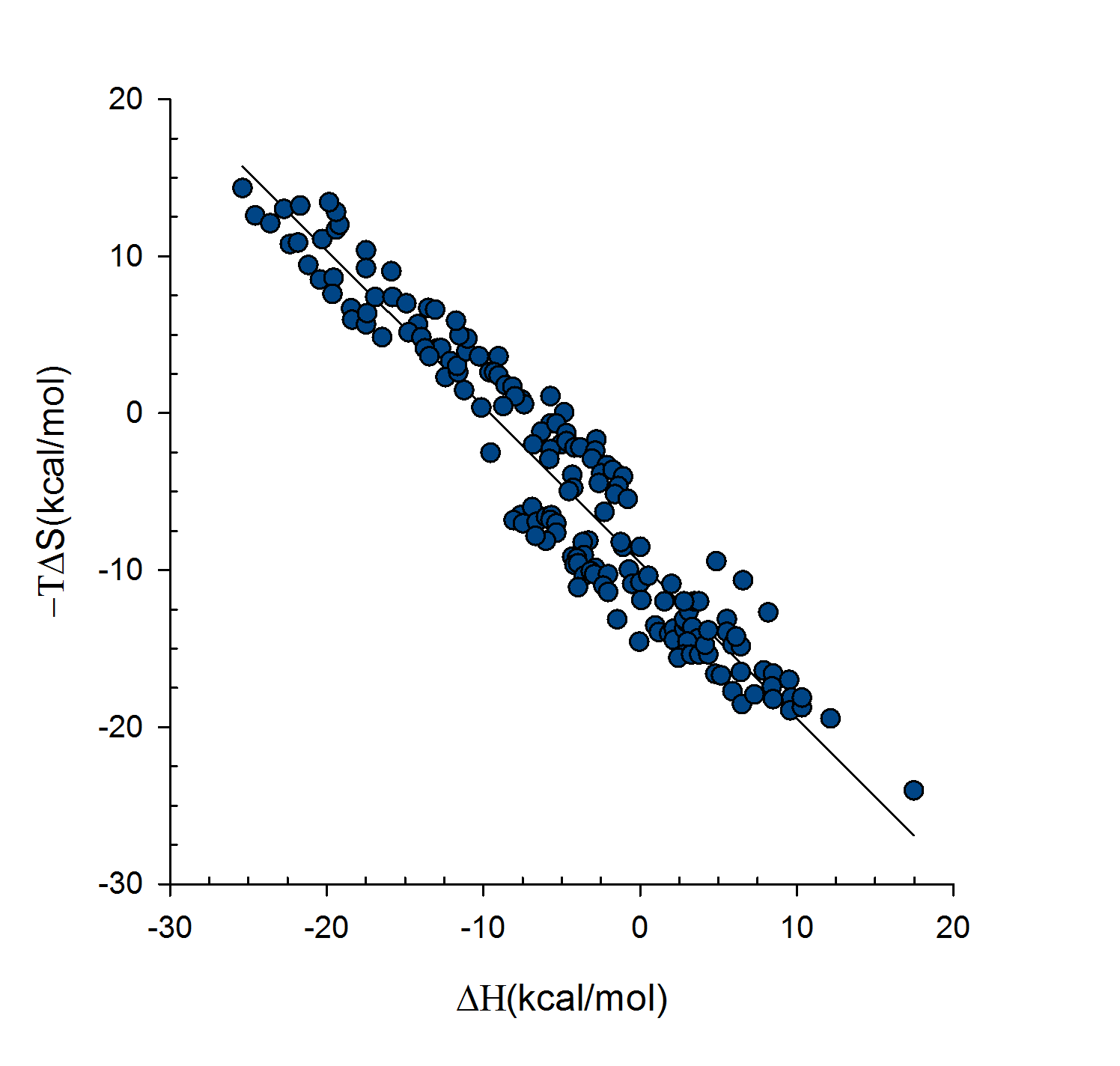
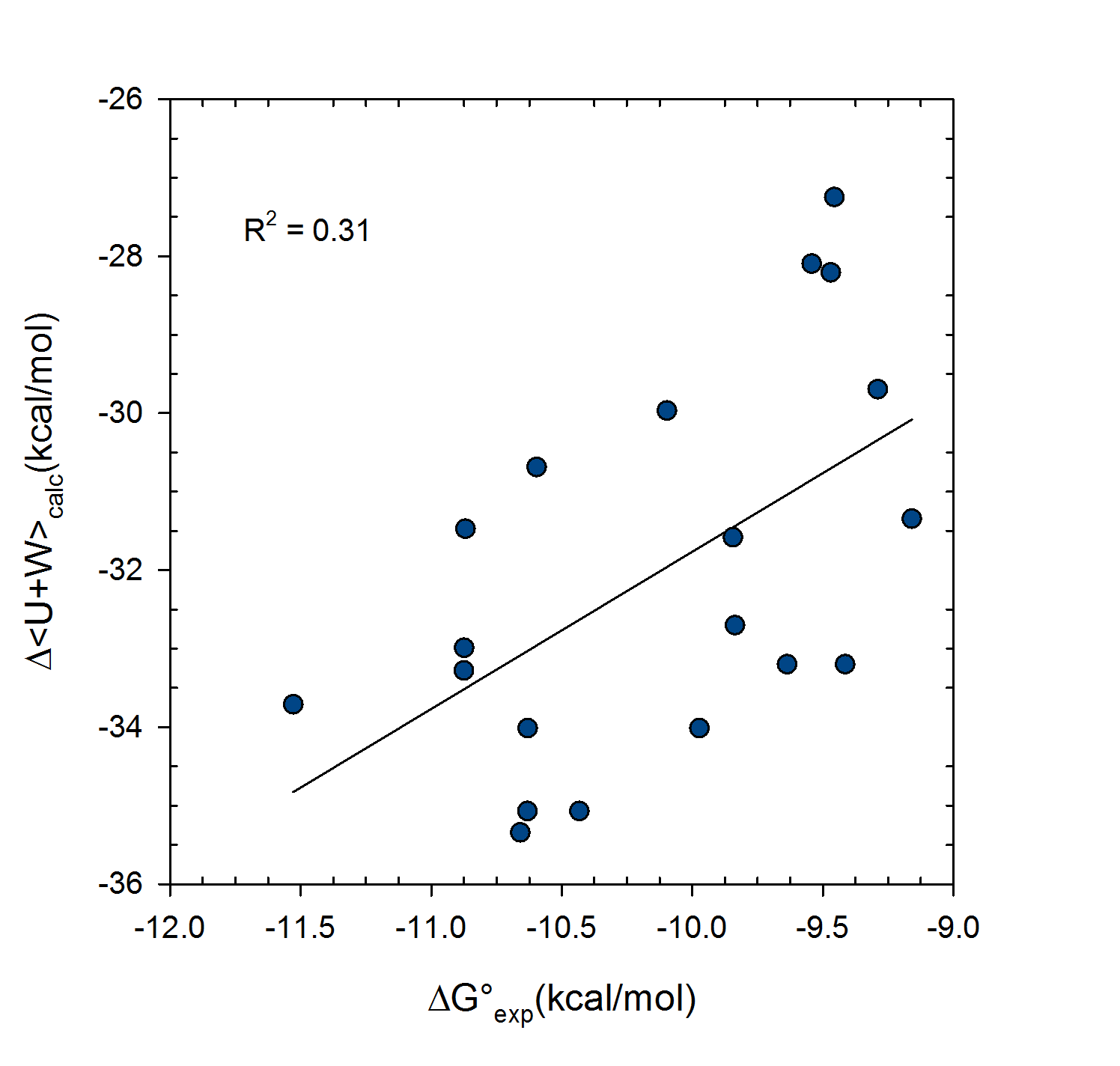
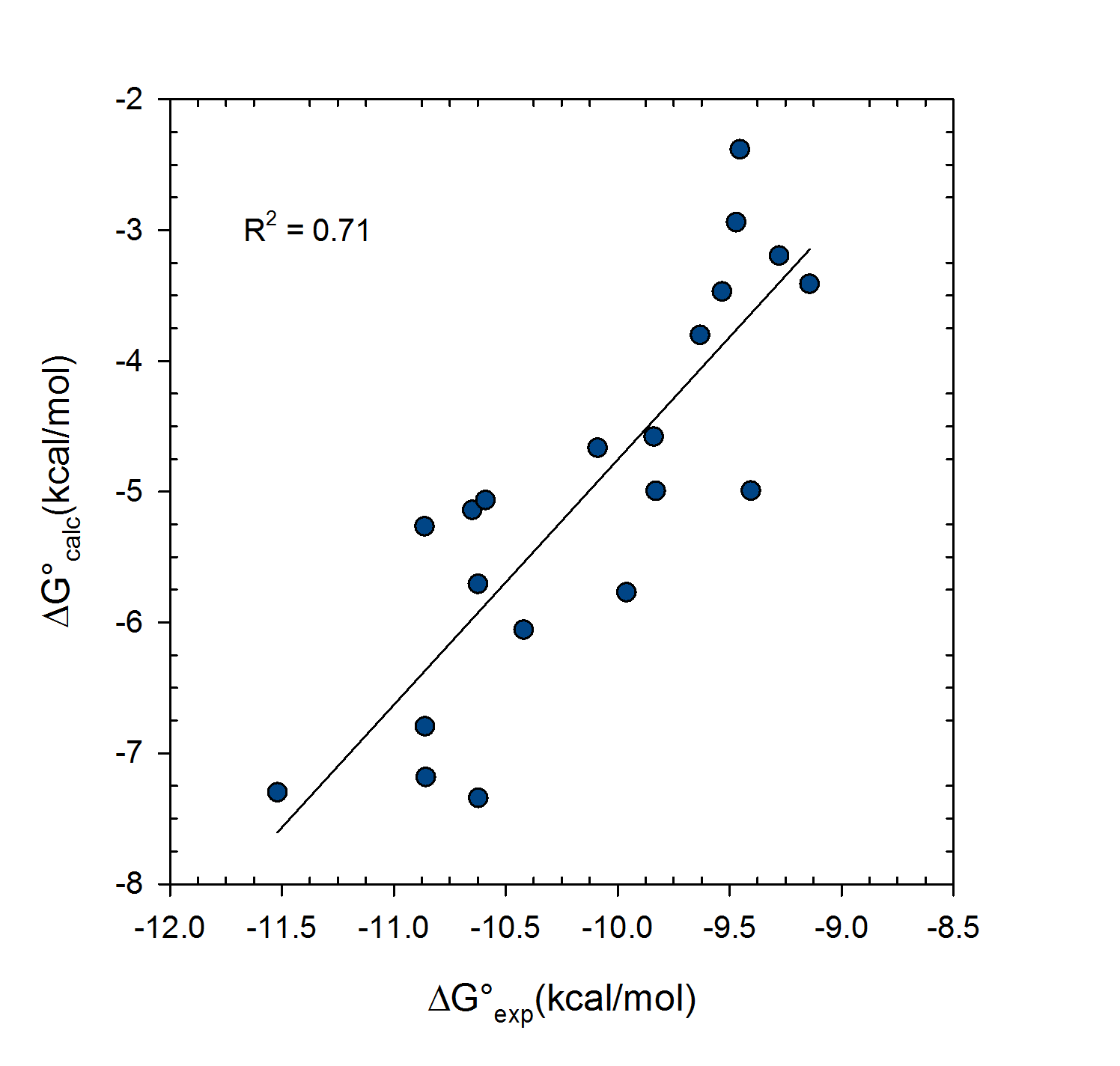
Statistical thermodynamics of binding
VM2 is solidly grounded in a statistical thermodynamics approach to the prediction of binding affinities. VM2 aims to predict standard free energies of binding. Experiments can determine standard binding free energies through the equilibrium constant Kbind (see below), where the equilibrium constant itself is determined via the receptor, ligand, receptor-ligand concentrations along with the standard concentration.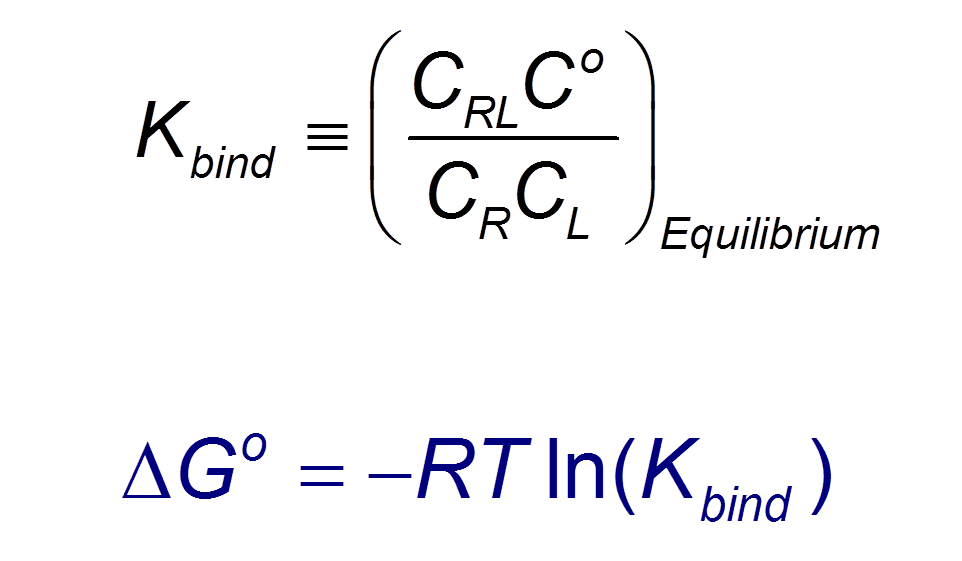
VM2 calculates the standard free energy of binding via the standard chemical potentials of the receptor, ligand, and receptor-ligand complex.
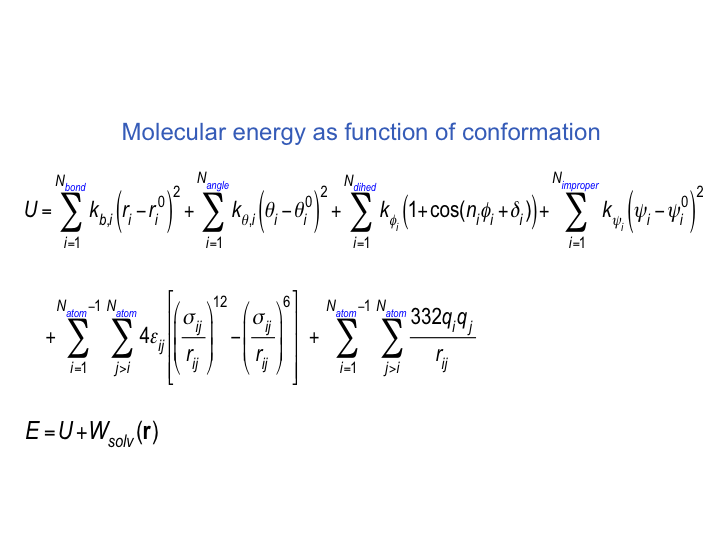
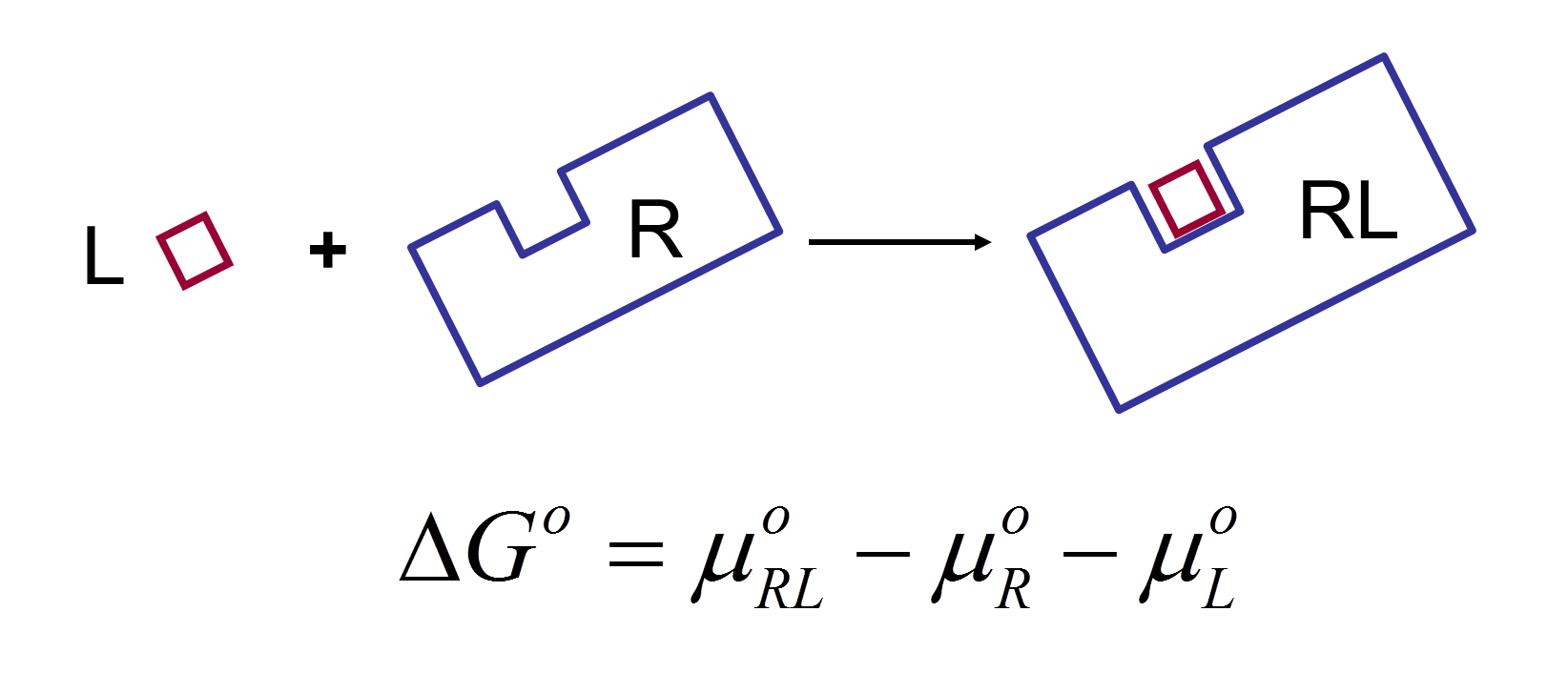 VM2 uses the classical formulation of the partition function for calculation of each chemical potential.
VM2 uses the classical formulation of the partition function for calculation of each chemical potential.

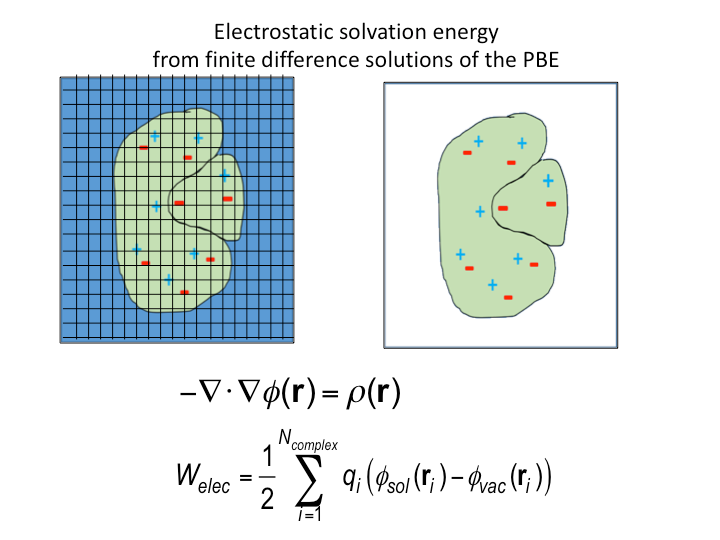
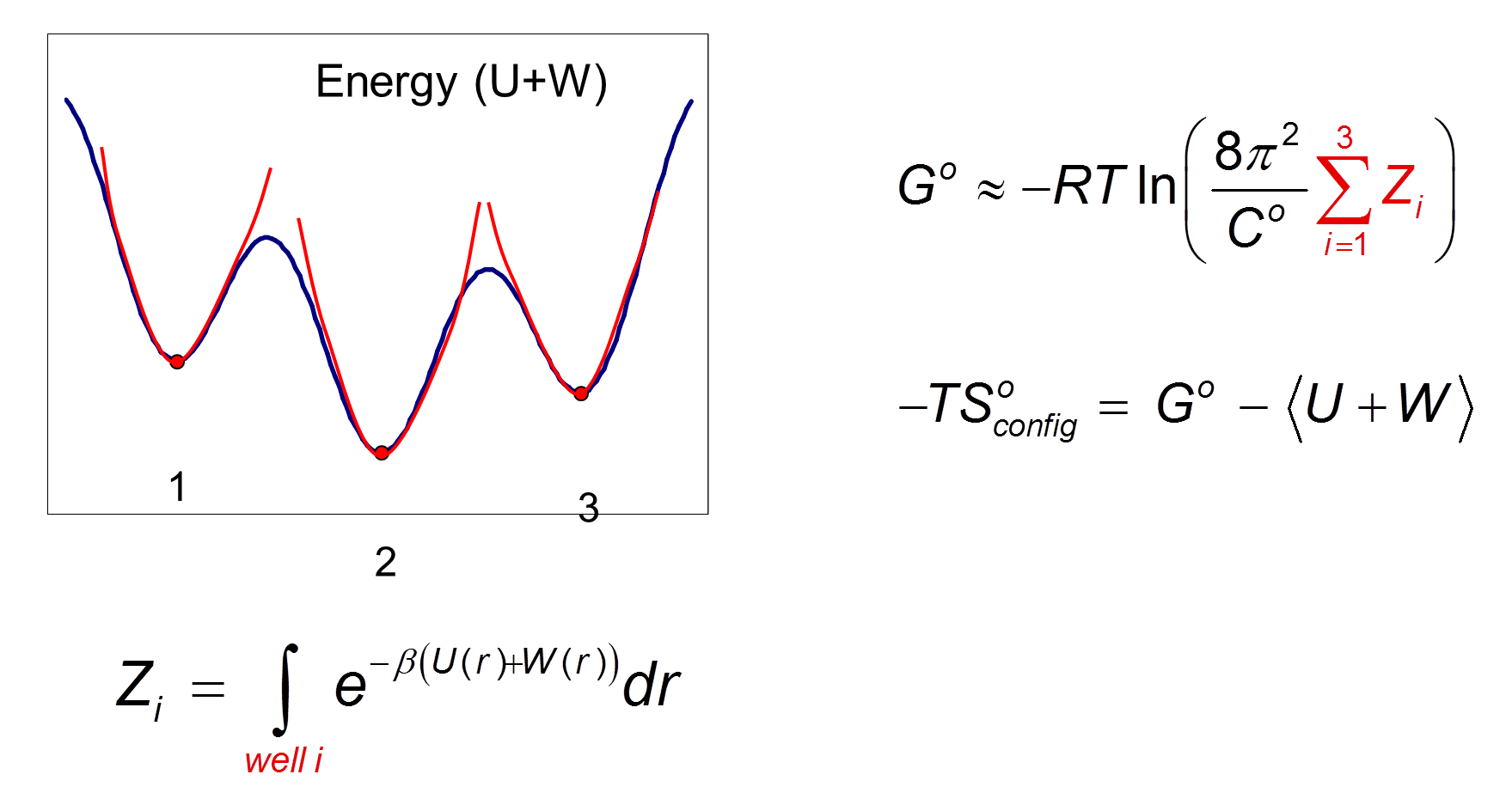
Here the external degrees of freedom have been integrated out and C0 provides a correction to the standard state. Formally, the configuration integral must be determined over all space along the remaining internal degrees of freedom, the VM2 method approximates this using the concept of mining minima.
Local configuration integrals
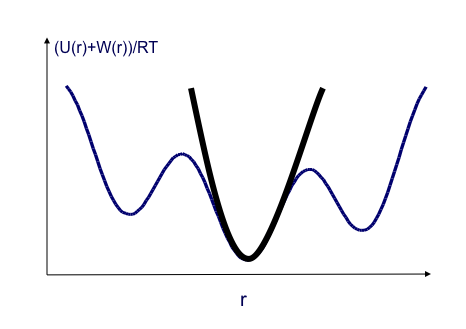
A Harmonic approximation together with a numeric correction for anharmonic low-energy modes, the Harmonic Approximation with Mode Scanning (HAMS) method, is used to calculate the local configuration integral
![]()
for each minimum produced. The harmonic approximation is applied via square roots of the Hessian matrix eigenvalues.
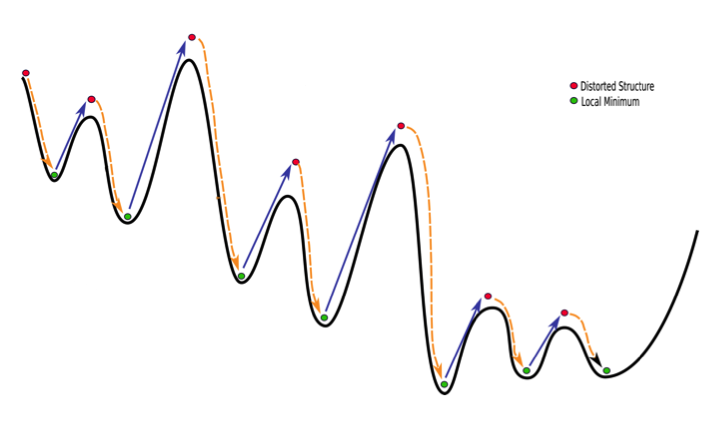
The Mode Scanning anharmonic corrections are calculated by numerical integration of the energy surfaces produced by distortion along low-energy internal-coordinate modes.
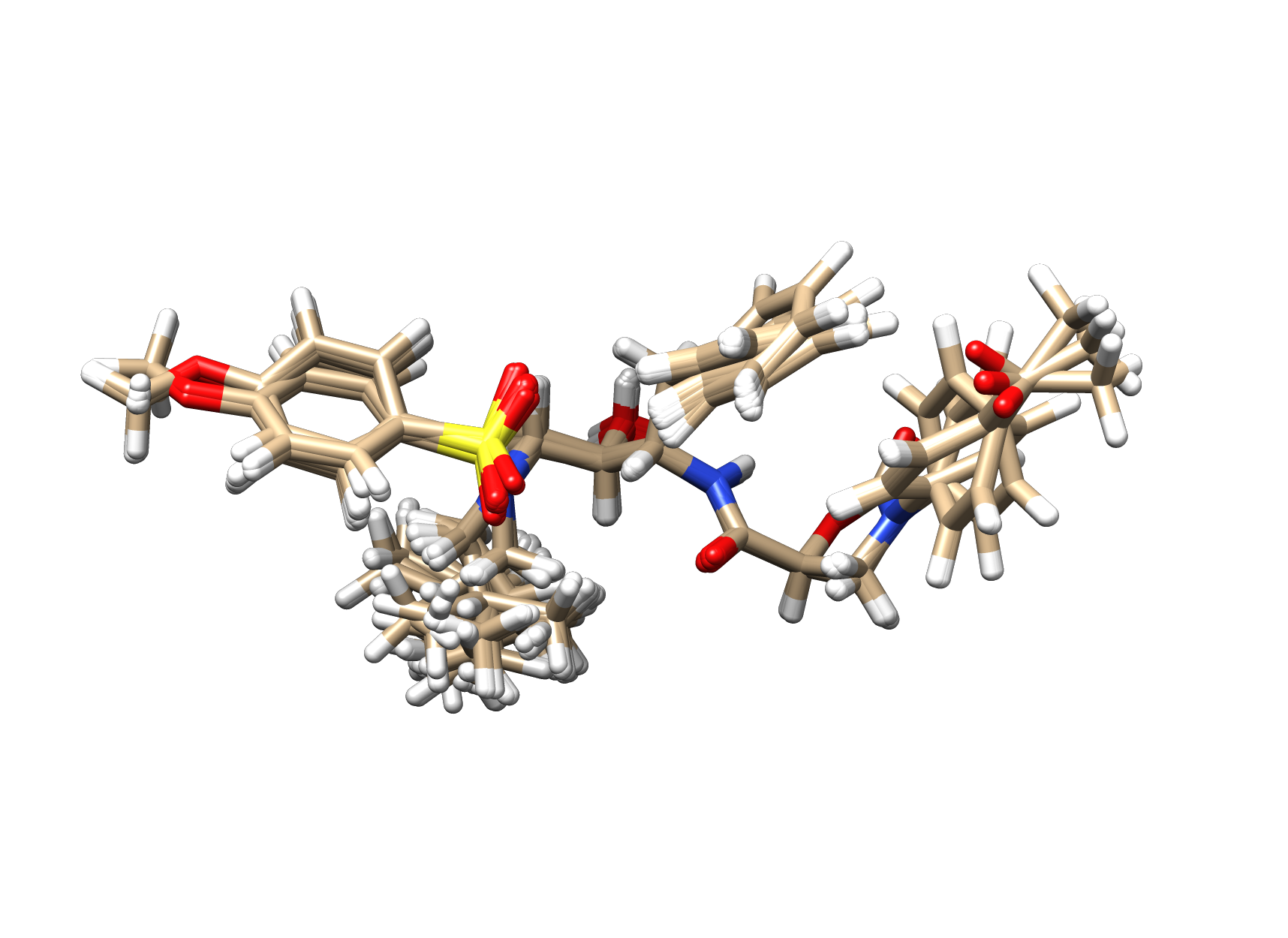
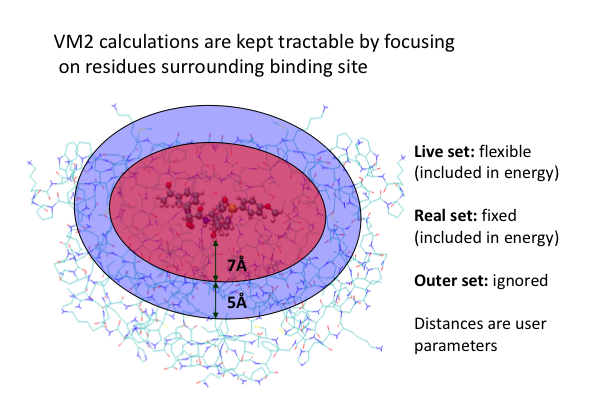
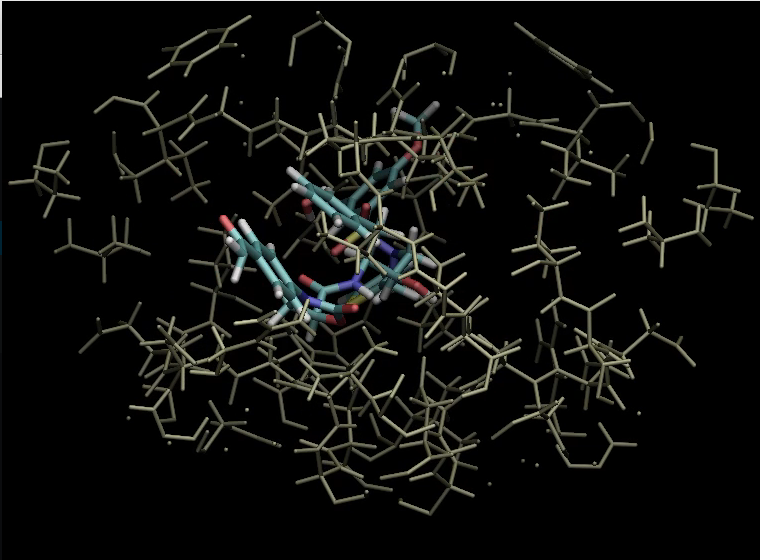
The following illustrates the case of a small molecule mode-distortion and minimization.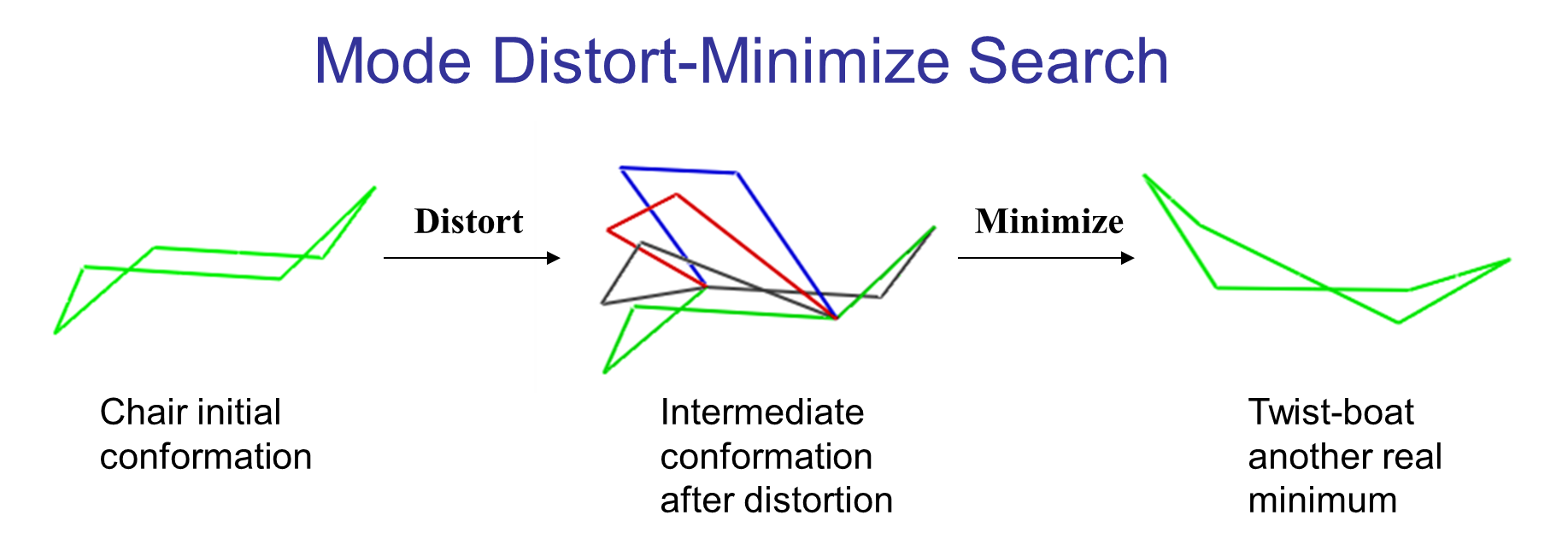 Each time a new lowest energy minimum is found it is subsequently used as a basis for the next mode distort-minimization. The number of torsional modes available depends on the size of the system.
Each time a new lowest energy minimum is found it is subsequently used as a basis for the next mode distort-minimization. The number of torsional modes available depends on the size of the system.
The search can cycle through the available modes in order (low to high energy) or at random. In addition, a linear combination of randomly chosen pairs of these modes can be used to define the distortion.
To further illustrate this approach consider the following six-atom system and its internal coordinates.
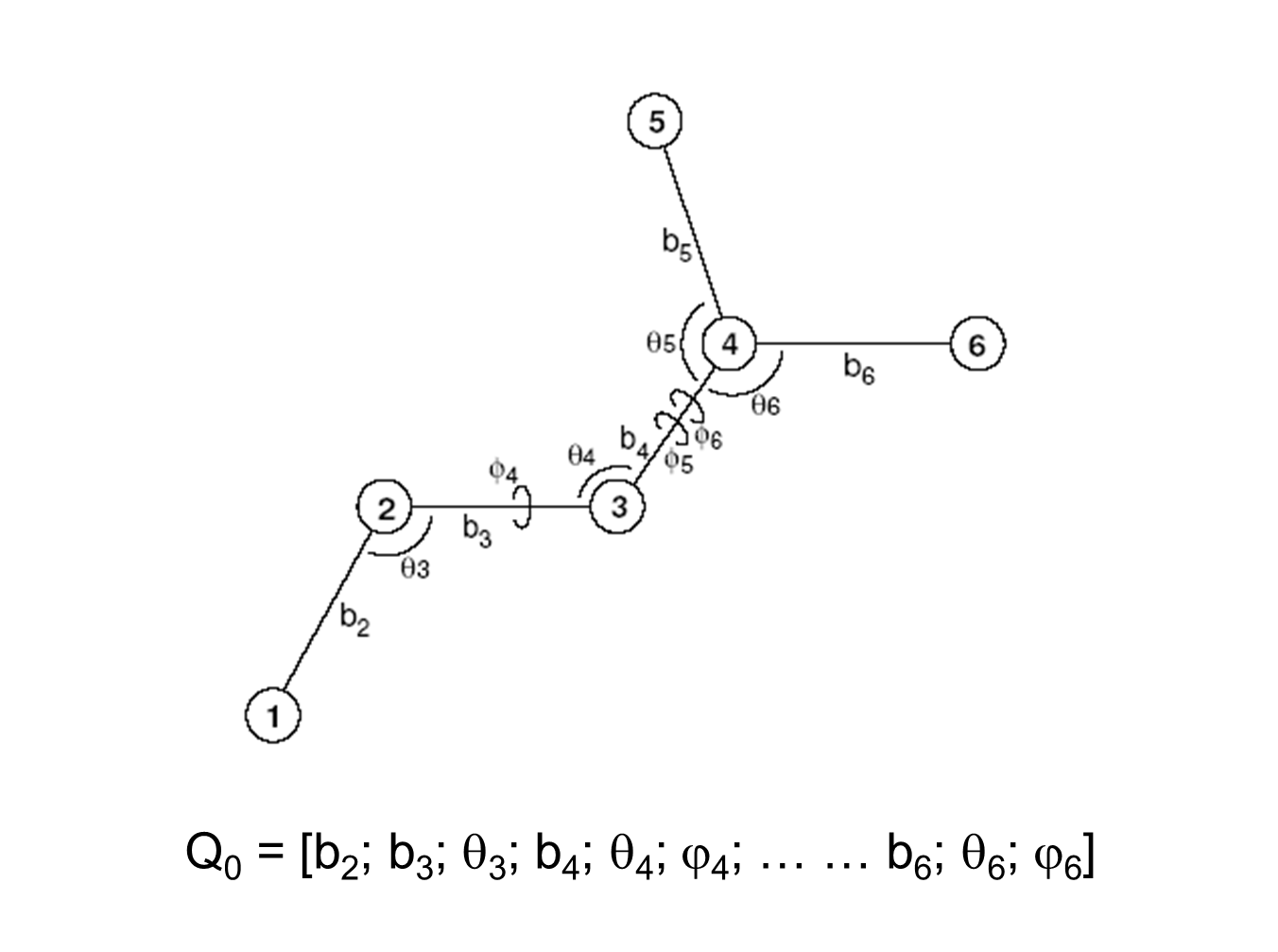 The total energy 2nd-derivative (Hessian) matrix with respect to internal coordinates (bond, angle, torsion coordinates) can be calculated
The total energy 2nd-derivative (Hessian) matrix with respect to internal coordinates (bond, angle, torsion coordinates) can be calculated
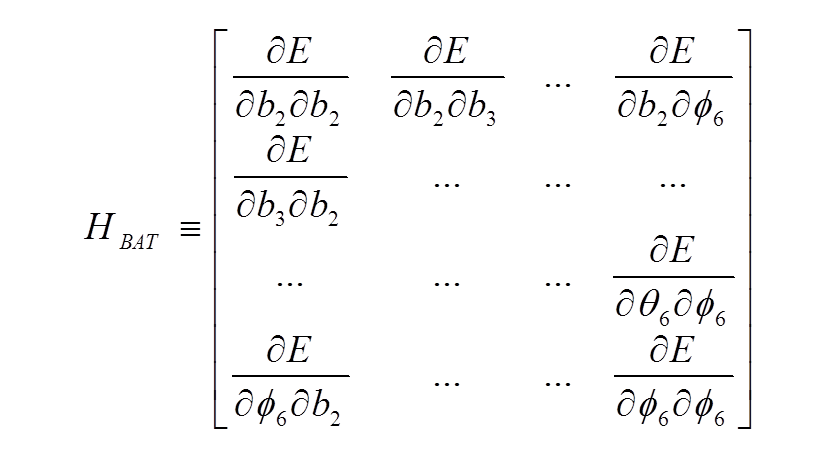
The dimensionality of the Hessian is reduced by only allowing rows and columns with dihedral angles; furthermore, only one dihedral angle per central bond is retained
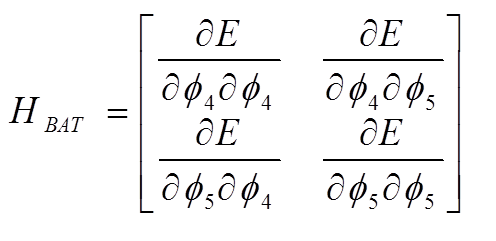
This reduced size Hessian is then diagonalized. The resulting eigenvectors are each a linear combination of the two dihedral “basis functions” and each represents a torsional mode distortion driver. The initial conformation can then be distorted along each driver D with step size α
![]()
and as previously described a quasi-Newton geometry optimzation carried out after the distortion is complete.
The following movies depict various types of mode distortions.