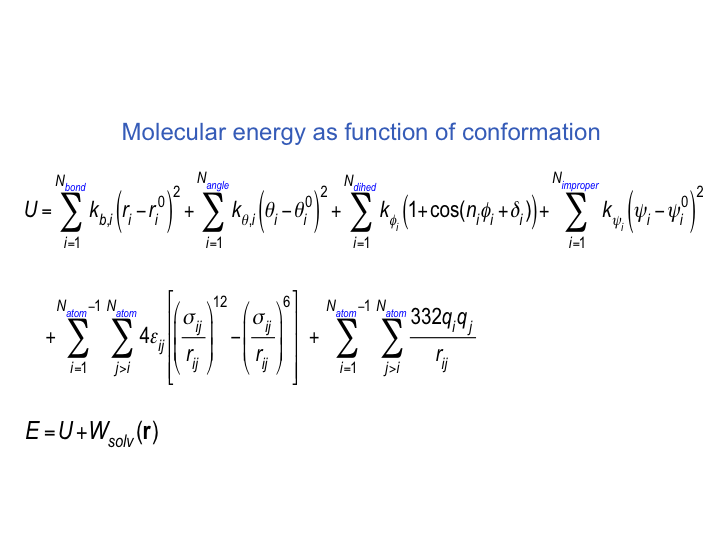
Currently for protein-ligand VM2 free energy calculations only classical molecular molecular mechanics force field underlying energy models are available.
Quantum mechanics
Development work is underway to interface VM2 with quantum mechanical potentials for application to small molecular systems such as ligand molecules and host-guest systems e.g. ligand complexes with cyclodextrin.