VCharge is an electronegativity equalization method, where the electronegativity of each atom depends upon its atomic number, hybridization, and bonding environment within the molecule, and where special constraints are applied to keep too much charge from flowing off ionized groups. The constrained equalization problem is solved by an adaptation of the method of Lagrangian multipliers.
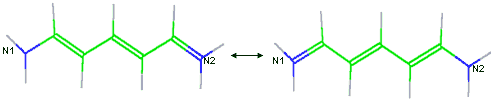
A unique feature of VCharge is its handling of alternate resonance forms. Alternate resonance forms of each molecule are automatically identified, and the atomic electronegativities and hardnesses are averaged over the resonance forms. This ensures that equal charges are assigned to chemically equivalent atoms, even when this equivalence is not apparent from the single resonance form provided in the input SDfile. For example, nitrogens N1 and N2 illustrated below would not be considered equivalent if resonance forms were not considered. Ionization and tautomer states are taken as specified in the input SDfile.