
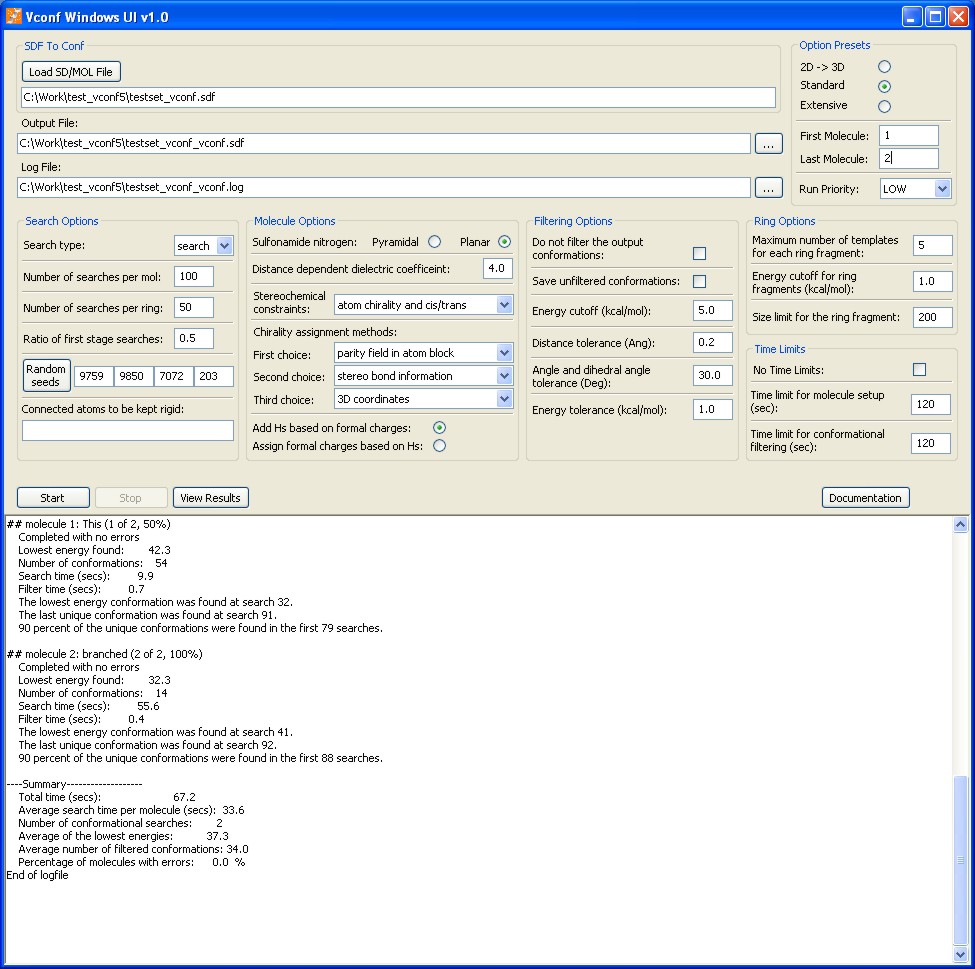
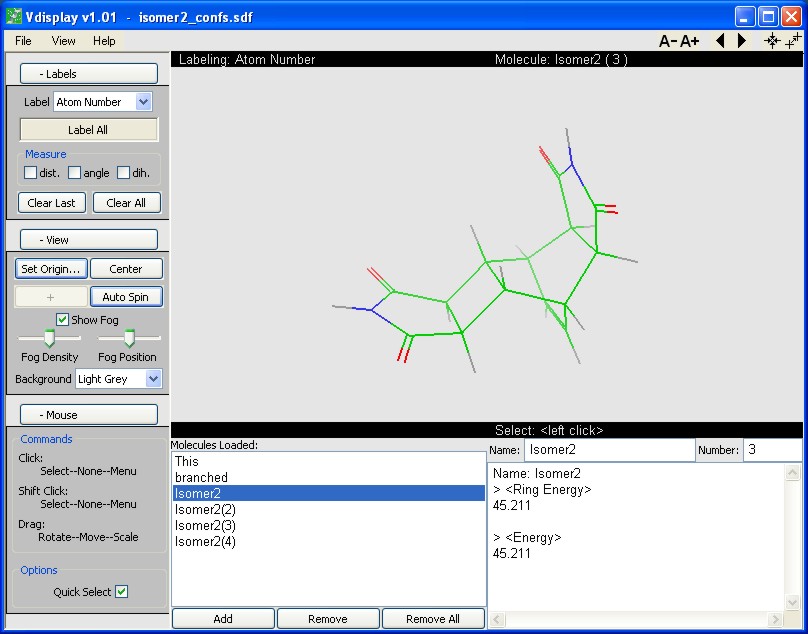
Vconf version 1.0 for Microsoft Windows® including a graphical user interface and Linux.

Vconf version 1.0 for Microsoft Windows® including a graphical user interface and Linux.
© 2007-2023 VeraChem, LLC
![]()