Home
Products
VM2
VDock
Small Molecule Tools >
VConf
VDraw
VMap
VCharge
VFilter
Vrms
VDisplay
News
About Us
Profile
Personnel
Contact
Support
Documentation
Licensing
Science
VM2 Method
Videos
Get VeraChem Software
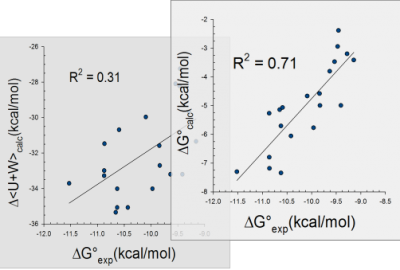
Computed binding free energies for ligand series ranking
True free energies calculated – entropy matters!
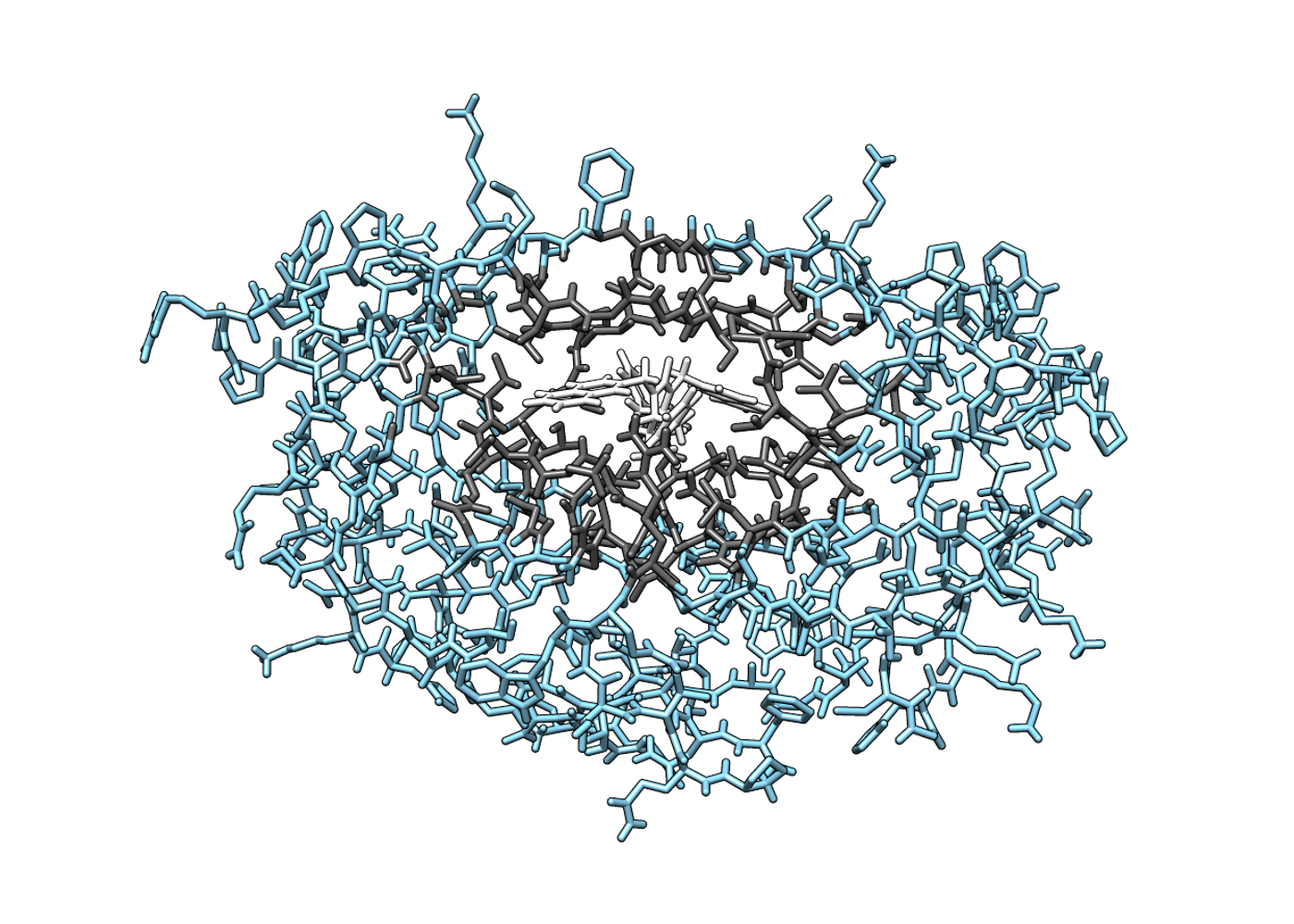
Fully flexible ligand and protein
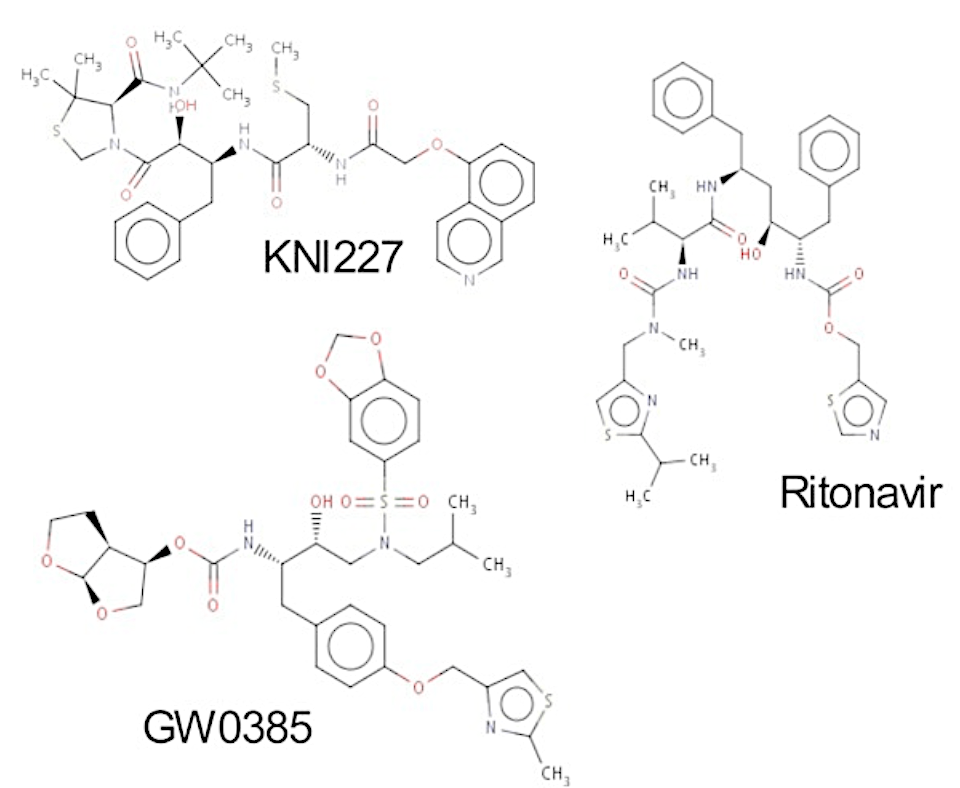
Handles diverse ligands with ease
Efficient and effective conformational search
Fast accurate solvation
Find all significant binding modes not just one
Understand your system through detailed energy decomposition and analysis
Easily access the power of the cloud
Fast multi-CPU and multi-GPU calculations
Multiple energy models
Flexible licensing for both commercial and academic use cases
Learn more
Get VM2
True free energies calculated
Free energy provides more accurate protein-ligand affinity prediction
Includes configurational and conformational entropy
Accounts for anharmonic effects
Learn More
Flexible ligand and protein
Fully mobile receptor region essential for accurate pose and affinity prediction
Protein backbone fully flexible as well as side chain
User can select regions to be mobile or fixed as required
Learn More
Handles diverse ligand sets
Unlike FEP methods arbitrary changes can easily be made to ligand functional groups
Can efficiently use system information if available e.g. pose
However can determine pose with no prior knowledge if required
Learn More
Highly efficient conformational searching
Conformational search engine efficiently and reliably finds low energy minima of large systems
Novel distort-minimize algorithms
Uses distortions along linear combinations of torsional modes
In addition rigid body rotation-translation searching
Learn More
Fast accurate solvation
Poisson-Boltzmann Surface Area (PBSA) corrected potential energies
Easily include explicit water molecules in the binding site when required
Learn More
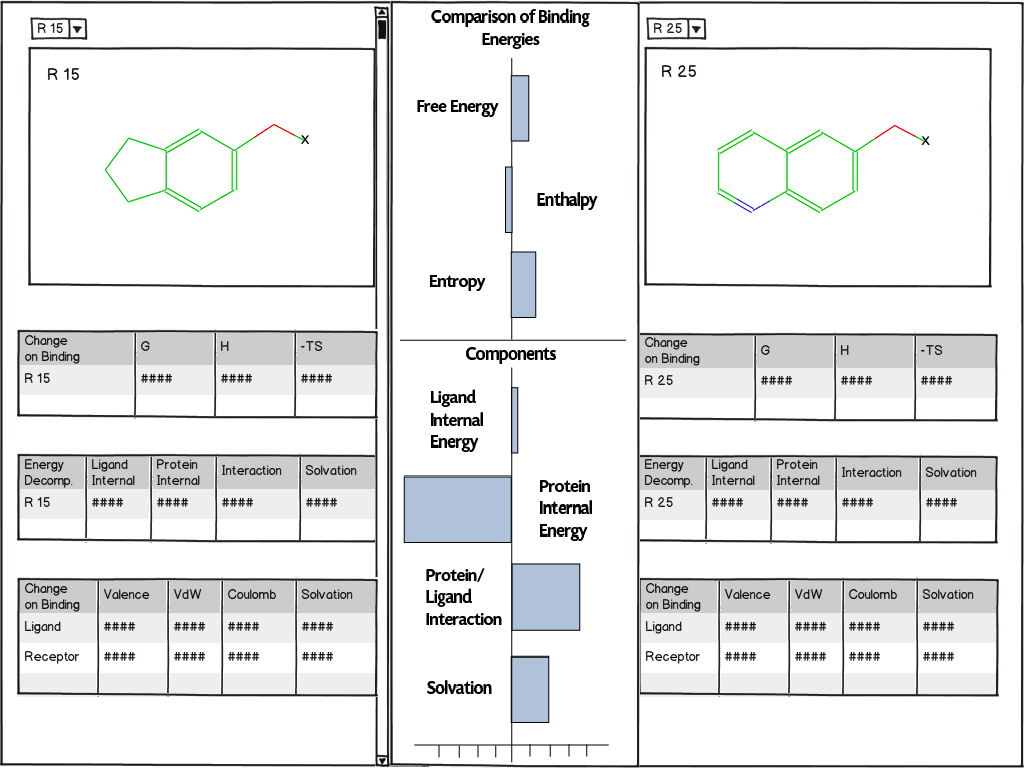
Energy decomposition for binding affinity analysis
Can determine favorable and unfavorable free energy contributions to the binding affinity of functional groups
Helps guide ligand design decisions in lead optimization efforts
Learn More
CloudVM2
Access the power of the Cloud
Ported to Amazon Web Services cloud platform and Google Cloud Platform
Easy to use graphical interface to submit VM2 calculations
Fast turnaround – calculate affinities for a whole protein-ligand series concurrently
Learn More
Get VM2