Cost per run an order of magnitude lower compared to a typical in-house compute cluster.
Graphical management console which can run on any desktop or in the cloud.
World class security, all calculations run behind a firewall managed by Amazon.
Compute power scaled to job size means results in days, not months.
CloudVM2 is an implementation of VeraChem’s VM2 molecular affinity binding prediction software on the Amazon Web Services cloud. It has been developed to allow researchers without access to a powerful computer cluster, or those who simply desire a more economical and convenient method to run large calculations, to use the novel VM2 method in their work. Additionally, CloudVM2 can provide results for a series of protein-ligand complexes much more quickly than all but the largest privately owned computer clusters.
For unbeatable cost effectiveness, CloudVM2 utilizes the Amazon Web Services spot market, where worldwide surplus computing power is auctioned at steep discounts averaging 80 percent. Amazon may shut down compute nodes available in the spot market at any time with a two minute warning. The CloudVM2 system has been developed to work with this unreliability via computation checkpointing and saving intermediate results in case of a shutdown. The computation may then be restarted from the last checkpoint when inexpensive surplus capacity again becomes available.
CloudVM2 User Interface
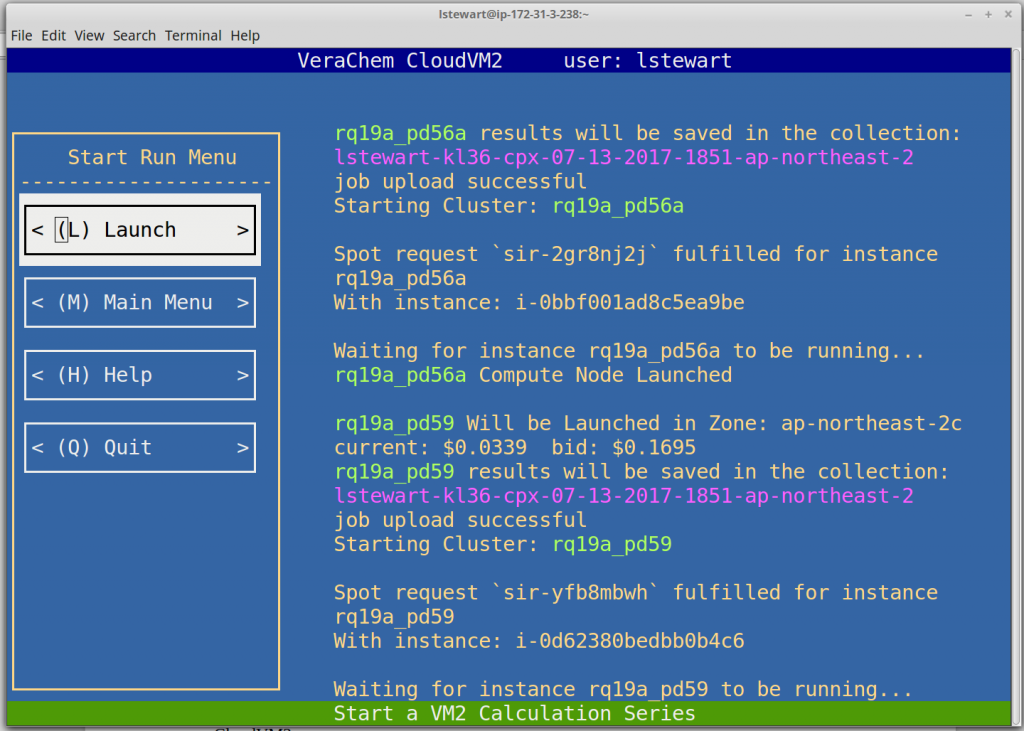
Launch Calculations
The CloudVM2 GUI is written as a terminal mode application, so it can be run on any operating system which supports the Python language and the GUI can also be accessed via ssh.
Launching a series of calculations is simple, the user enters the path to the directory containing their series and then clicks . CloudVM2 will then retrieve recent spot price history from all data centers worldwide, select the most advantageous region, and start uploading the calculations. Current spot prices are retrieved after each compute node launch and as prices rise due to the demand CloudVM2 is placing on the spot market, calculations may get load-balanced across the AWS regions.
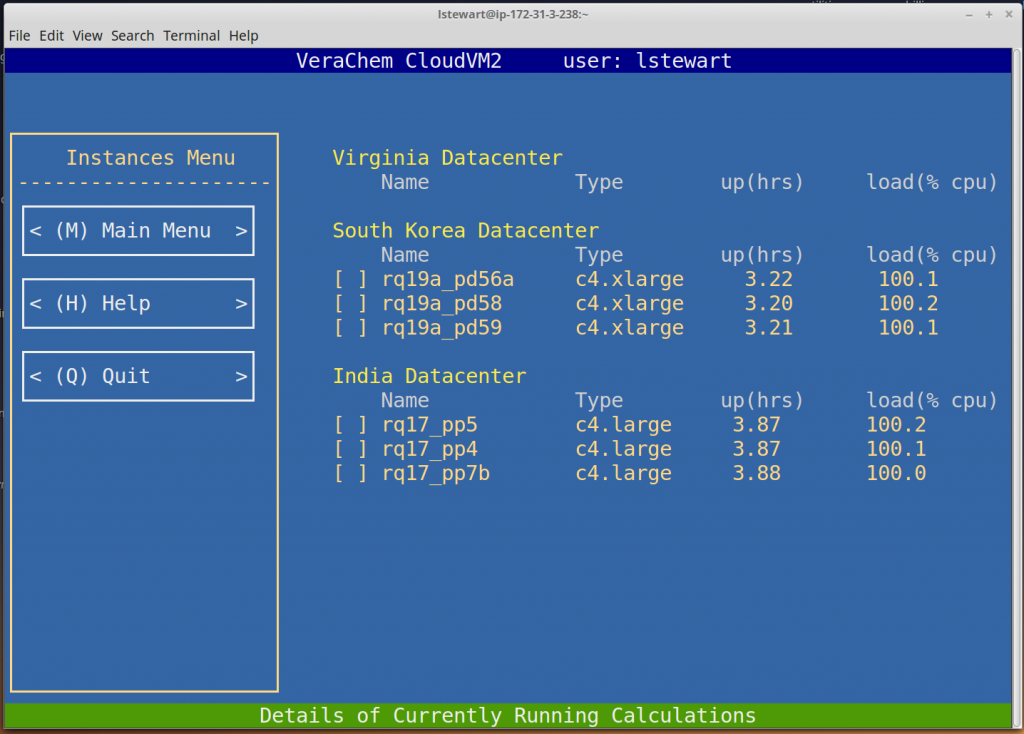
Monitor Compute Nodes
CloudVM2 allows the user to monitor calculations in progress, displaying each active compute node, sorted by region. Should a problem arise the user can terminate a compute node by selecting it and clicking .
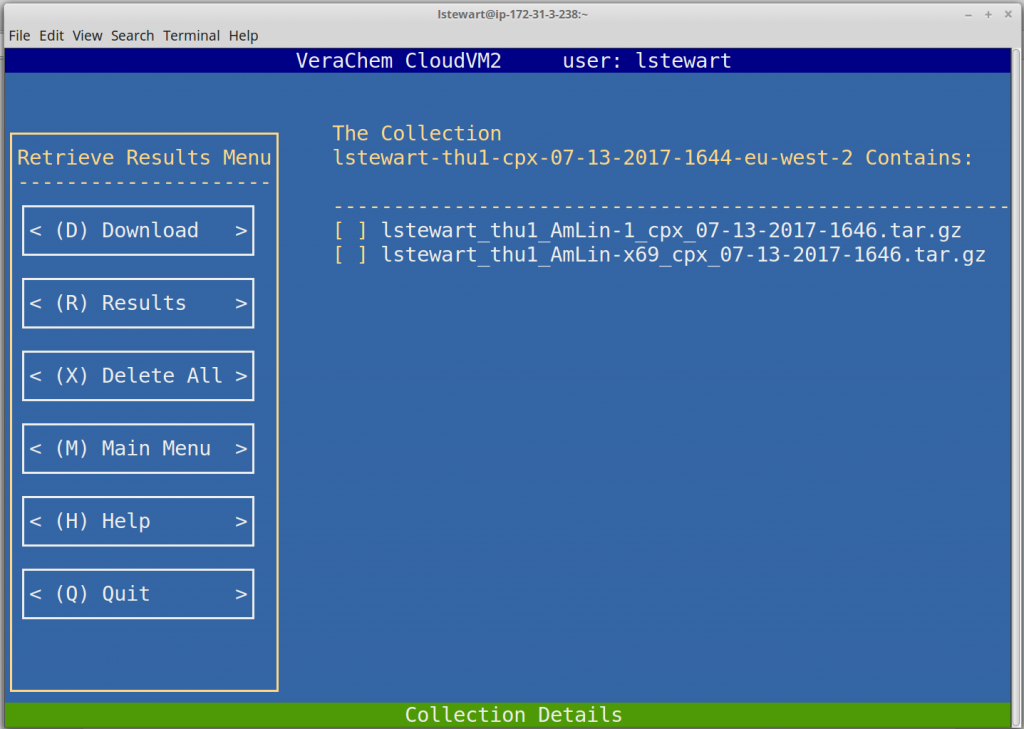
Retrieve Results
CloudVM2 utilizes Amazon’s Simple Storage Service (S3) for persistent data storage. Completed calculation results and intermediate restart files are transferred to S3, where the user can download them at their convenience.